

MINERVA: A Facile Strategy for SARS-CoV-2 Whole-Genome Deep Sequencing of Clinical Samples
- PMID: 33290743
- PMCID: PMC7678446
- DOI: 10.1016/j.molcel.2020.11.030
MINERVA: A Facile Strategy for SARS-CoV-2 Whole-Genome Deep Sequencing of Clinical Samples
Abstract
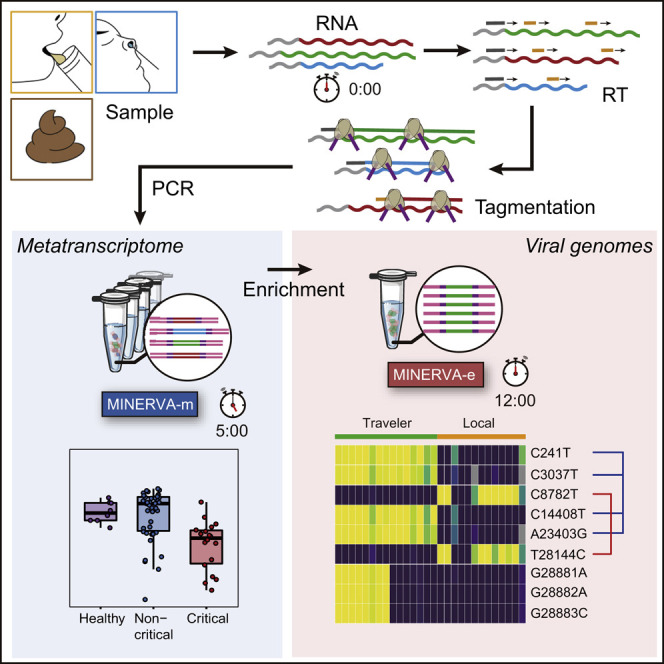
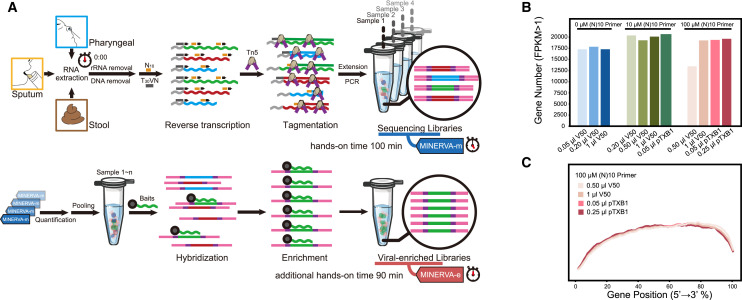
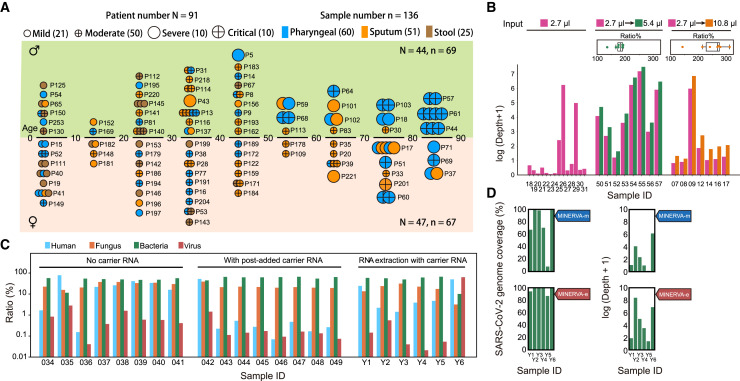
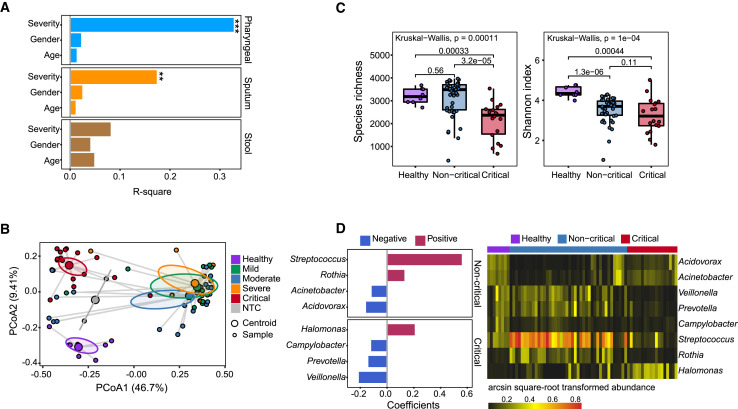
Analyzing the genome of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) from clinical samples is crucial for understanding viral spread and evolution as well as for vaccine development. Existing RNA sequencing methods are demanding on user technique and time and, thus, not ideal for time-sensitive clinical samples; these methods are also not optimized for high performance on viral genomes. We developed a facile, practical, and robust approach for metagenomic and deep viral sequencing from clinical samples. We demonstrate the utility of our approach on pharyngeal, sputum, and stool samples collected from coronavirus disease 2019 (COVID-19) patients, successfully obtaining whole metatranscriptomes and complete high-depth, high-coverage SARS-CoV-2 genomes with high yield and robustness. With a shortened hands-on time from sample to virus-enriched sequencing-ready library, this rapid, versatile, and clinic-friendly approach will facilitate molecular epidemiology studies during current and future outbreaks.
Keywords: COVID-19, SARS-CoV-2, whole-genome sequencing, metagenomics.
Copyright © 2020 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of Interests The authors declare no competing interests.
Figures
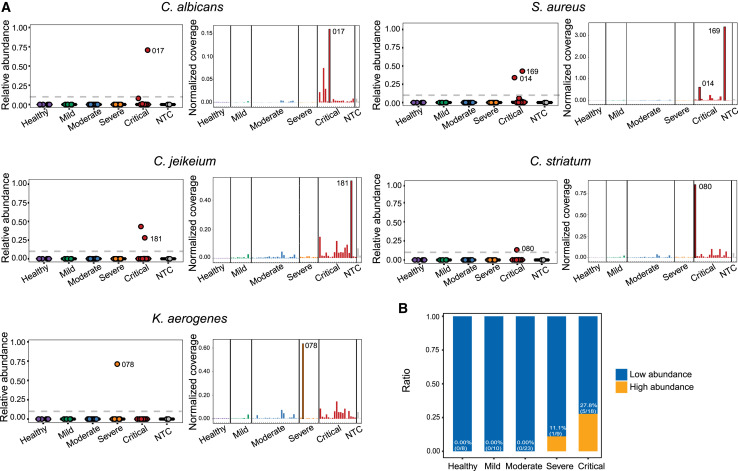
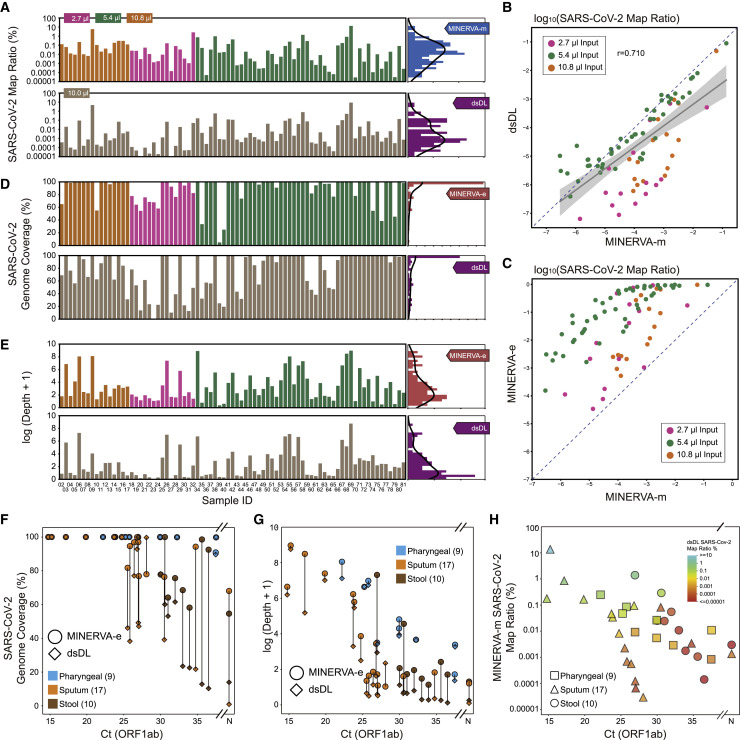
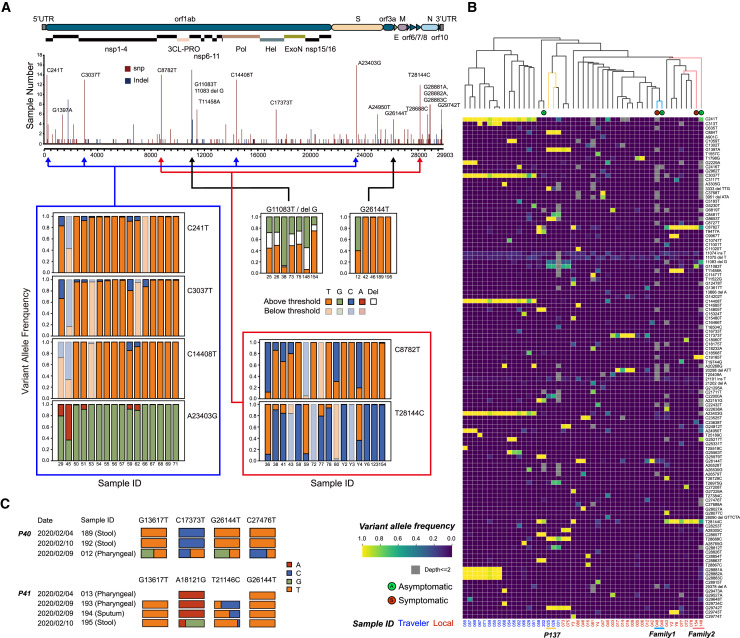
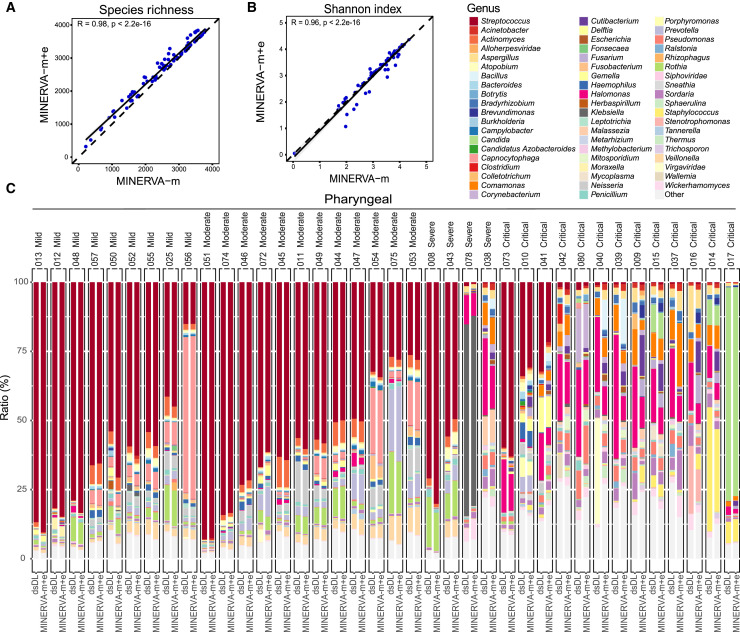
References
-
- Bushnell B. Accurate, Splice-Aware Aligner; 2014. BBMap: A Fast.
-
- Carey V.J. Ported to R by Thomas Lumley (versions 3.13, 4.4, version 4.13). R package version. 2006;4:13. B. R. gee: Generalized Estimation Equation solver. https://CRAN.R-project.org/package=gee. 11.
-
- Carl G., Kühn I. Analyzing spatial autocorrelation in species distributions using Gaussian and logit models. Ecol. Modell. 2007;207:159–170.
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
