

Type 1 diabetes mellitus as a disease of the β-cell (do not blame the immune system?)
- PMID: 33293704
- PMCID: PMC7722981
- DOI: 10.1038/s41574-020-00443-4
Type 1 diabetes mellitus as a disease of the β-cell (do not blame the immune system?)
Abstract
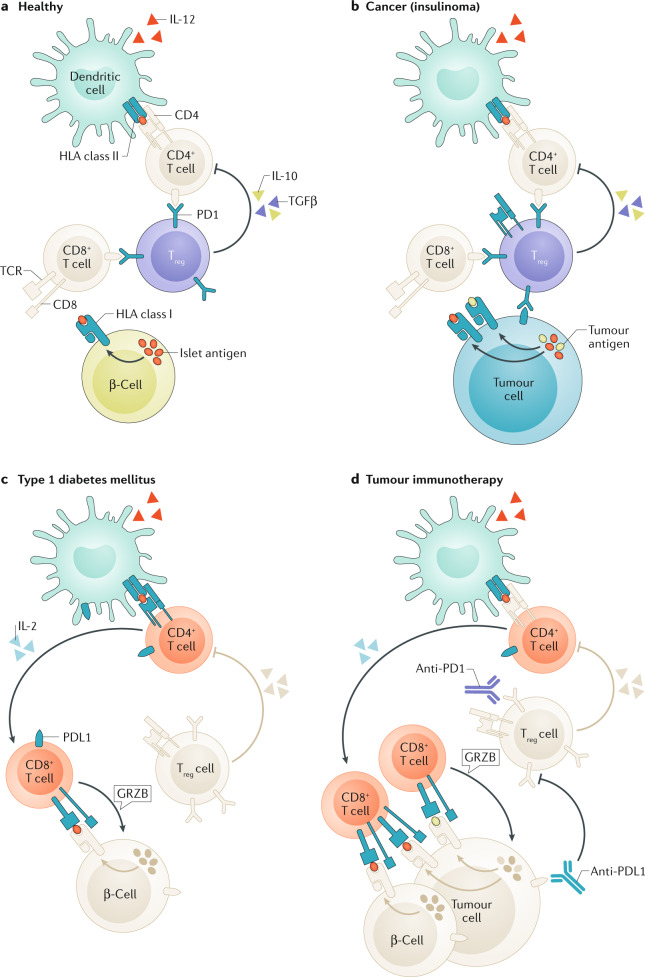
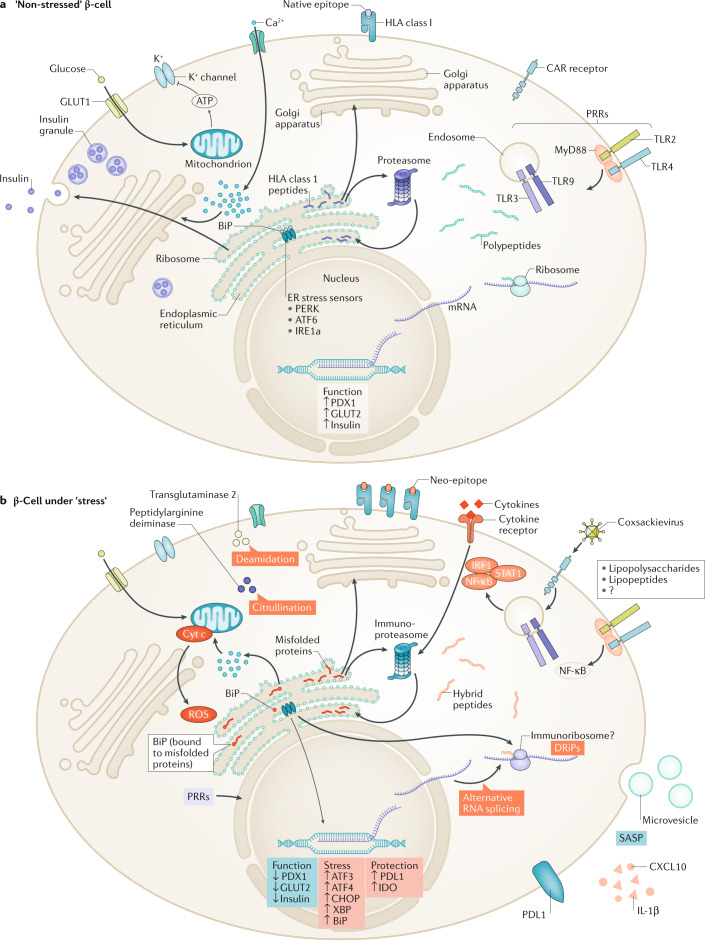
Type 1 diabetes mellitus is believed to result from destruction of the insulin-producing β-cells in pancreatic islets that is mediated by autoimmune mechanisms. The classic view is that autoreactive T cells mistakenly destroy healthy ('innocent') β-cells. We propose an alternative view in which the β-cell is the key contributor to the disease. By their nature and function, β-cells are prone to biosynthetic stress with limited measures for self-defence. β-Cell stress provokes an immune attack that has considerable negative effects on the source of a vital hormone. This view would explain why immunotherapy at best delays progression of type 1 diabetes mellitus and points to opportunities to use therapies that revitalize β-cells, in combination with immune intervention strategies, to reverse the disease. We present the case that dysfunction occurs in both the immune system and β-cells, which provokes further dysfunction, and present the evidence leading to the consensus that islet autoimmunity is an essential component in the pathogenesis of type 1 diabetes mellitus. Next, we build the case for the β-cell as the trigger of an autoimmune response, supported by analogies in cancer and antitumour immunity. Finally, we synthesize a model ('connecting the dots') in which both β-cell stress and islet autoimmunity can be harnessed as targets for intervention strategies.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- Gepts W. Islet changes suggesting a possible immune aetiology of human diabetes mellitus. Acta Endocrinol. Suppl. 1976;205:95–106. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
