

Combining structure and genomics to understand antimicrobial resistance
- PMID: 33294134
- PMCID: PMC7683289
- DOI: 10.1016/j.csbj.2020.10.017
Combining structure and genomics to understand antimicrobial resistance
Abstract
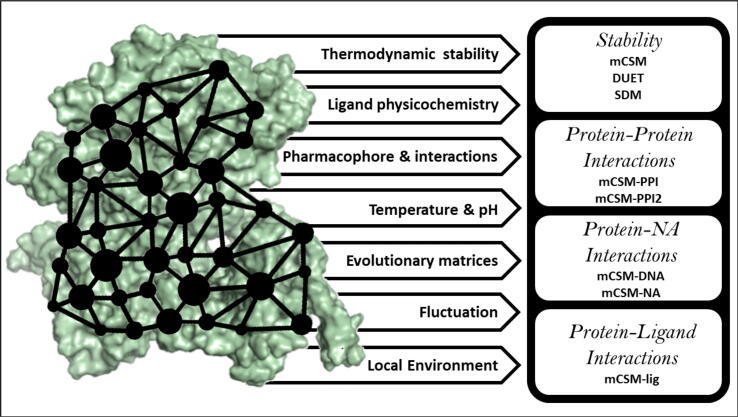
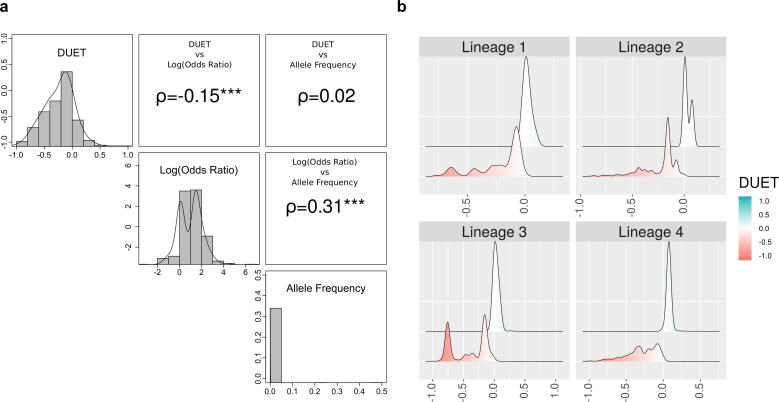
Antimicrobials against bacterial, viral and parasitic pathogens have transformed human and animal health. Nevertheless, their widespread use (and misuse) has led to the emergence of antimicrobial resistance (AMR) which poses a potentially catastrophic threat to public health and animal husbandry. There are several routes, both intrinsic and acquired, by which AMR can develop. One major route is through non-synonymous single nucleotide polymorphisms (nsSNPs) in coding regions. Large scale genomic studies using high-throughput sequencing data have provided powerful new ways to rapidly detect and respond to such genetic mutations linked to AMR. However, these studies are limited in their mechanistic insight. Computational tools can rapidly and inexpensively evaluate the effect of mutations on protein function and evolution. Subsequent insights can then inform experimental studies, and direct existing or new computational methods. Here we review a range of sequence and structure-based computational tools, focussing on tools successfully used to investigate mutational effect on drug targets in clinically important pathogens, particularly Mycobacterium tuberculosis. Combining genomic results with the biophysical effects of mutations can help reveal the molecular basis and consequences of resistance development. Furthermore, we summarise how the application of such a mechanistic understanding of drug resistance can be applied to limit the impact of AMR.
Keywords: Antimicrobial resistance; Genome wide association studies; Machine learning; Pathogen surveillance; Structural bioinformatics; Tuberculosis.
© 2020 The Authors.
Conflict of interest statement
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Figures
References
-
- WHO AMR-Fact-Sheet. WHO | 10 Facts on Antimicrobial Resistance 2018. https://www.who.int/news-room/facts-in-pictures/detail/antimicrobial-res... [accessed October 3, 2019].
-
- O’Neill Commission. Tackling Drug-Resistant Infections Globally-Final Report and Recommendations. The Review on Antimicrobial Resistance, Chaired by Jim O’Neill. 2016.
-
- WHO. Global tuberculosis report 2018; 2018.
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources