

Molecular docking, molecular dynamics, and in vitro studies reveal the potential of angiotensin II receptor blockers to inhibit the COVID-19 main protease
- PMID: 33294721
- PMCID: PMC7713577
- DOI: 10.1016/j.heliyon.2020.e05641
Molecular docking, molecular dynamics, and in vitro studies reveal the potential of angiotensin II receptor blockers to inhibit the COVID-19 main protease
Abstract
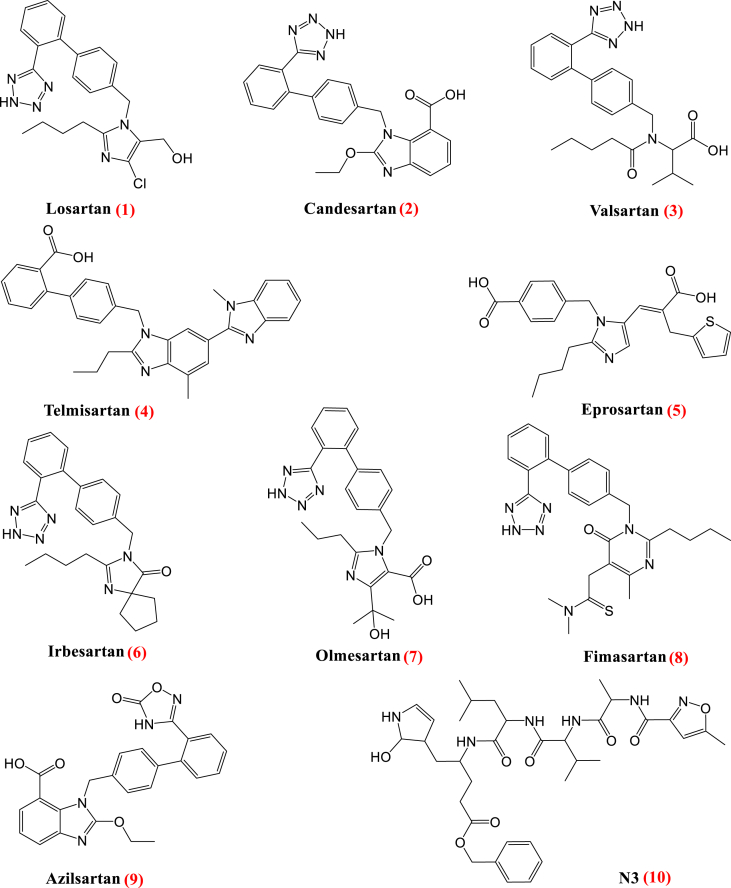
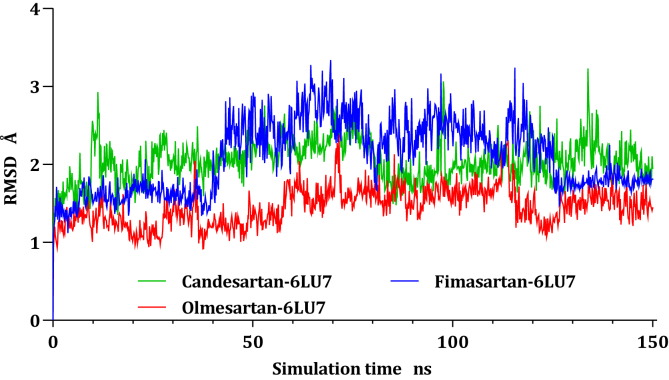
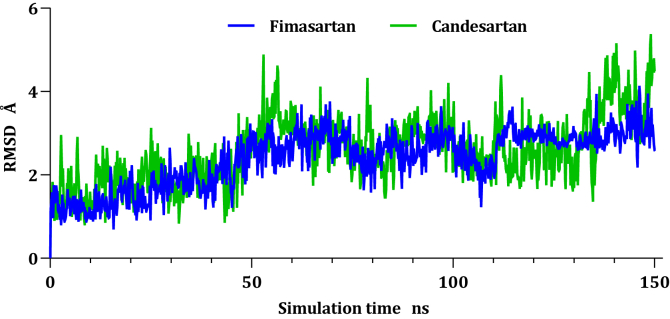
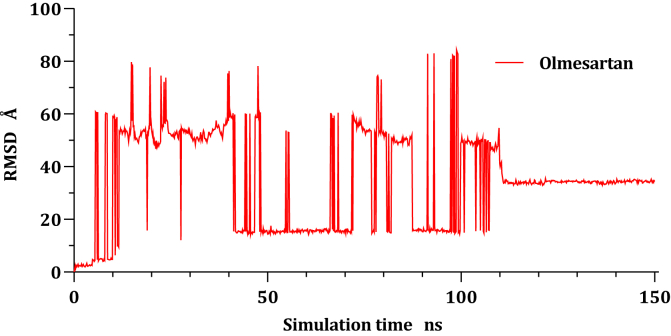
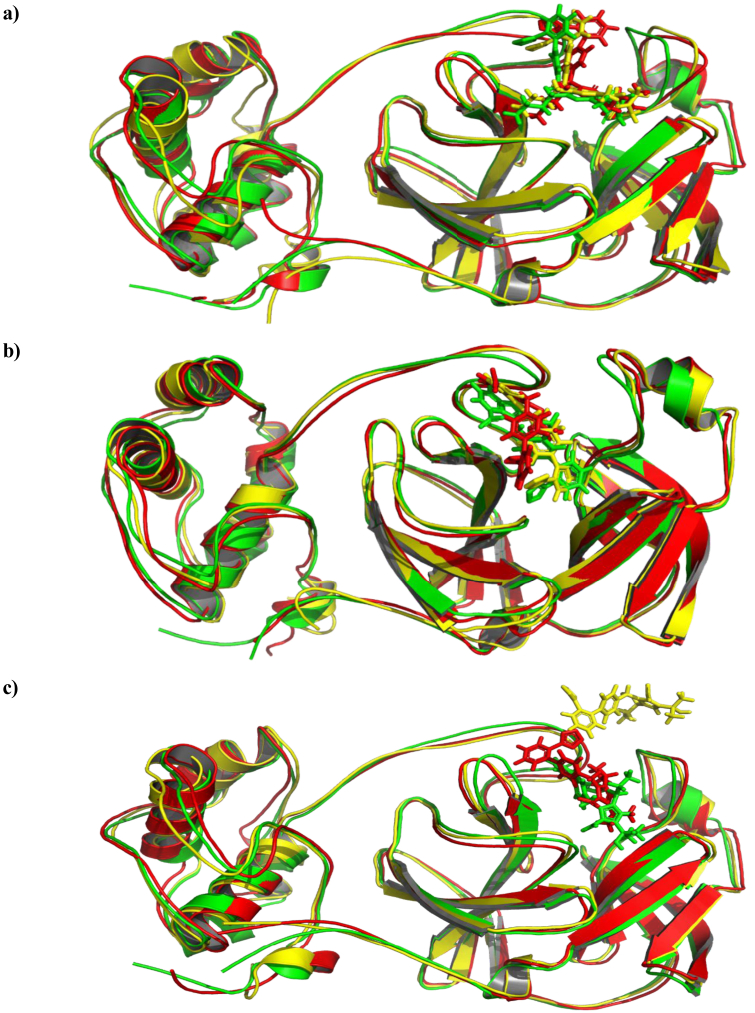
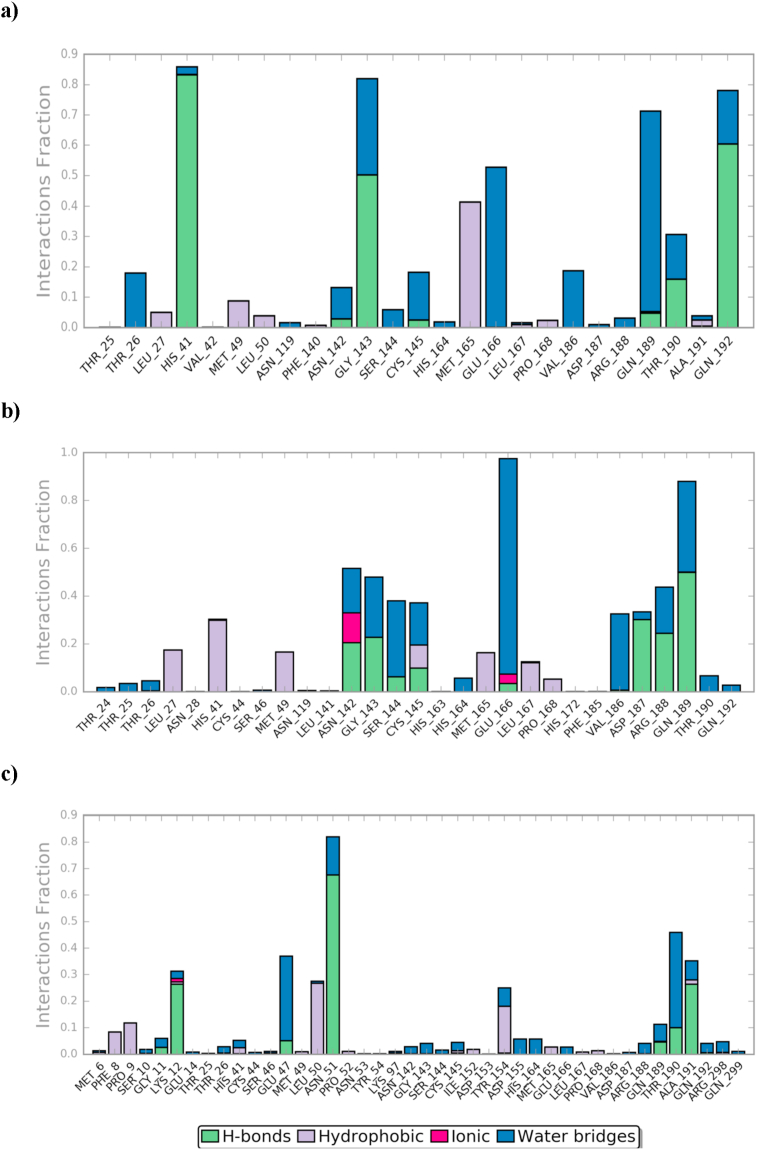
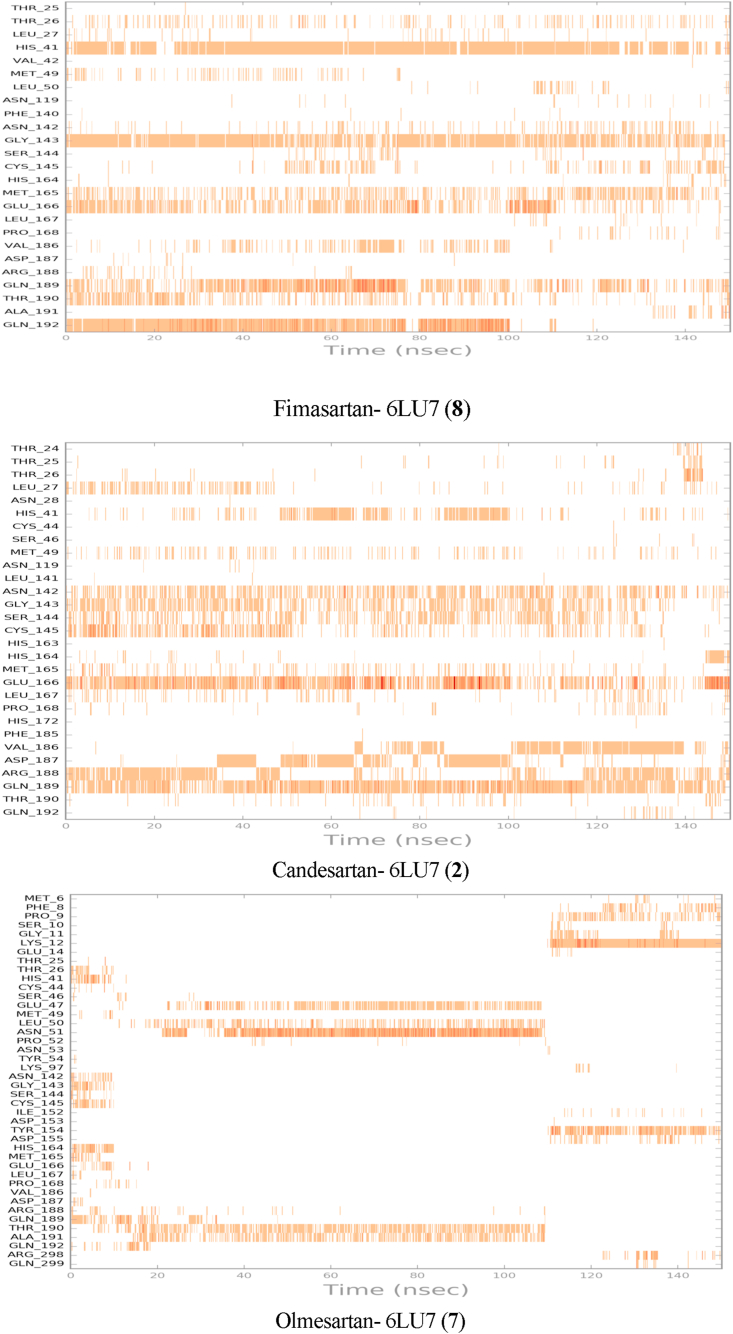
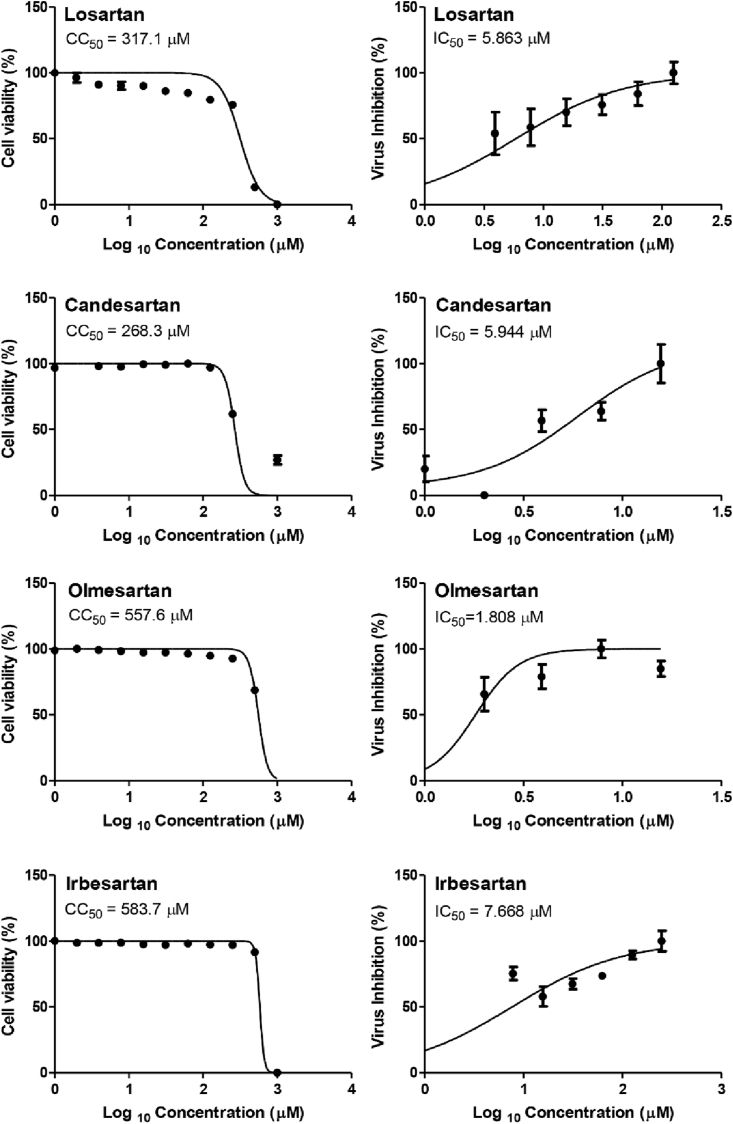
Drug repurposing is the most rapid and economic way nowadays to rapidly provide effective drugs for our pandemic coronavirus disease 2019 (COVID-19). It was a great debate about ARBs whether to be stopped or continued for patients using them especially at the beginning of the COVID-19 pandemic. In this study, we carried out a virtual screening for almost all members of ARBs (nine) against COVID-19 main protease. Molecular docking as one of the important computational techniques was performed in this work. Interestingly, the tested compounds showed variable binding affinities in the order of N3 inhibitor (10, docked) > Fimasartan (8) > Candesartan (2) > Olmesartan (7) > Azilsartan (9) > Eprosartan (5) > Valsartan (3) > Losartan (1) > Irbesartan (6) > Telmisartan (4). Moreover, Fimasartan (8), Candesartan (2), and Olmesartan (7) were additionally estimated through molecular dynamic simulations monitored via computing the binding free energy using MM-GBSA. The results are promising for rapidly repurposing such drugs (especially, Fimasartan (8) and Candesartan (2)) after further preclinical and clinical studies either alone or in combination with others for the treatment of COVID-19 virus especially known to cause vasodilatation (to prevent blood coagulation) and to reduce inflammation and fibrosis (to prevent pulmonary fibrosis), with well-known safety profiles. In vitro, the virtual findings were consistent with the experimental testing of four representative ARBs. Out of the tested compounds, Olmesartan (7) showed the most promising anti-SARS-CoV-2 activity (IC50 = 1.808 μM, and CC50 = 557.6 μM) with high selectivity index (308.4) against SARS-CoV-2 in Vero E6 cells. This work may clarify and approve not only the safety of ARBs used by a large group of patients worldwide but also their possible effectiveness against the COVID-19 virus either as a prophylactic or treatment option. It intended also to give a clear spot on the structure-activity relationship (SAR) required for the future design of new drugs targeting the newly emerged SARS-CoV-2 protease by medicinal chemists.
Keywords: ARBs; Biological sciences; COVID-19; Chemistry; Computer science; Docking; Drug repurposing; In vitro studies; MD; MM-GBSA; Materials science; Physics.
© 2020 The Author(s).
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Gautret P., Lagier J., Parola P. Department of virology , biological and pathological center , centre hospitalier. Int. J. Antimicrob. Agents. 2020:105949. - PubMed
-
- WHO . 2020. Coronavirus Disease (COVID-19) Pandemic.https://www.who.int/emergencies/diseases/novel-coronavirus-2019 - PubMed
-
- Mali S.N. The rise of new coronavirus infection-(COVID-19): a recent update. Eur. J. Med. Oncol. 2020;4(1):35–41.
LinkOut - more resources
Full Text Sources
Miscellaneous
