

Overcoming Adaptive Resistance to KRAS and MEK Inhibitors by Co-targeting mTORC1/2 Complexes in Pancreatic Cancer
- PMID: 33294856
- PMCID: PMC7691443
- DOI: 10.1016/j.xcrm.2020.100131
Overcoming Adaptive Resistance to KRAS and MEK Inhibitors by Co-targeting mTORC1/2 Complexes in Pancreatic Cancer
Abstract
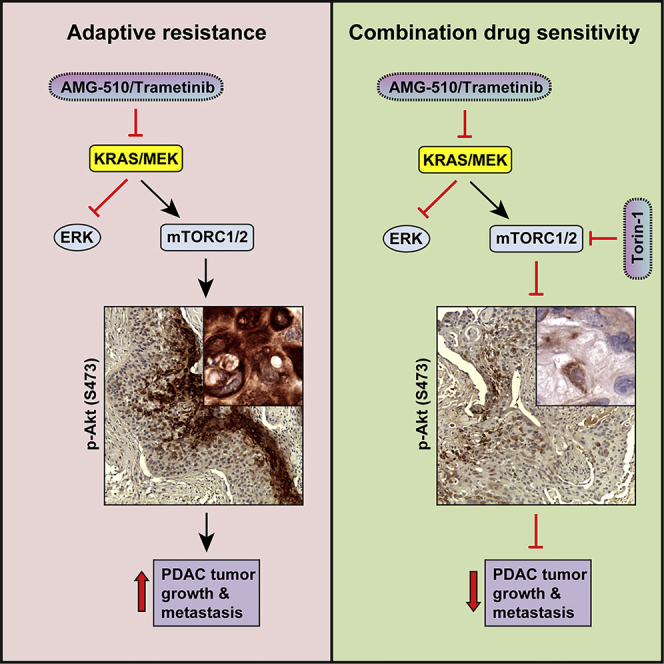
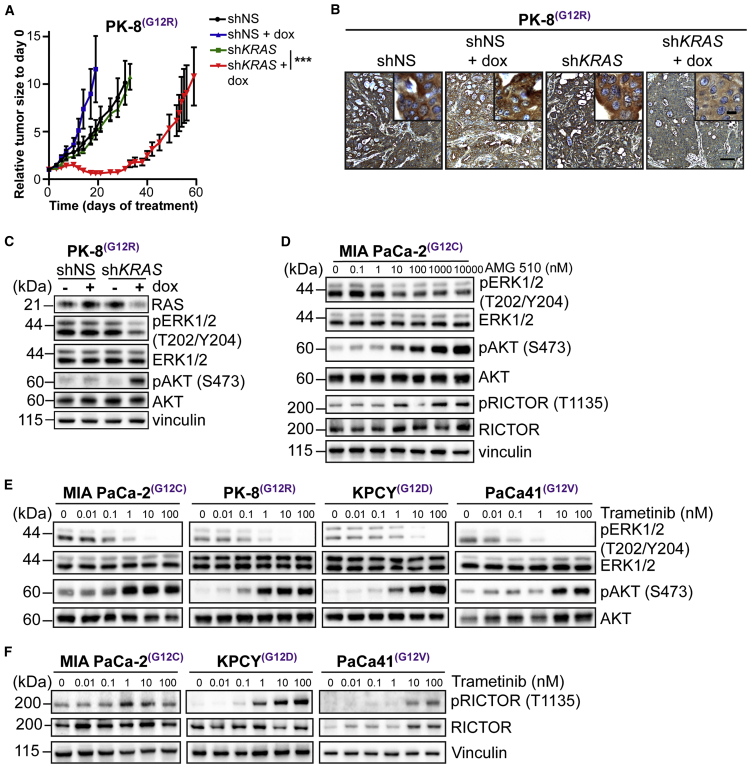
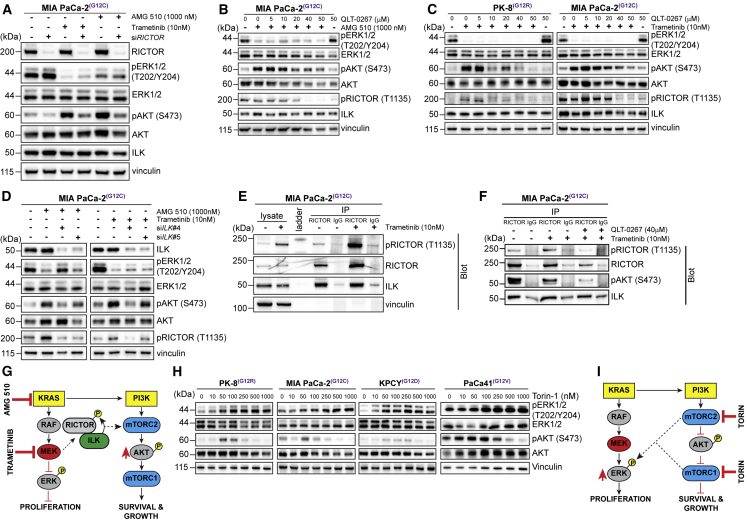
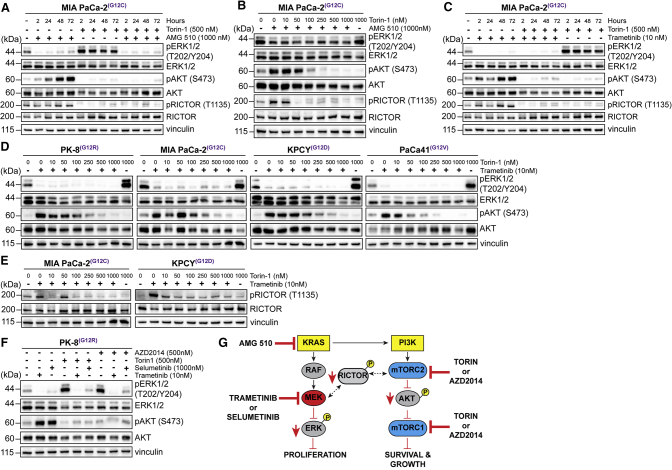
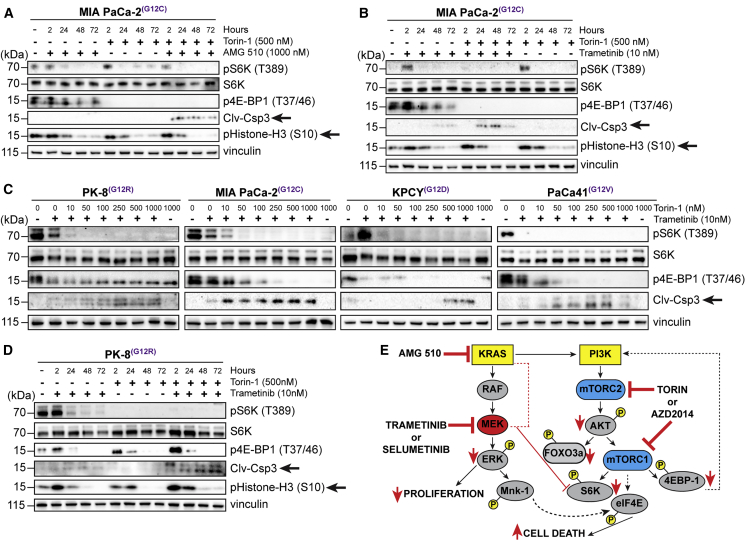
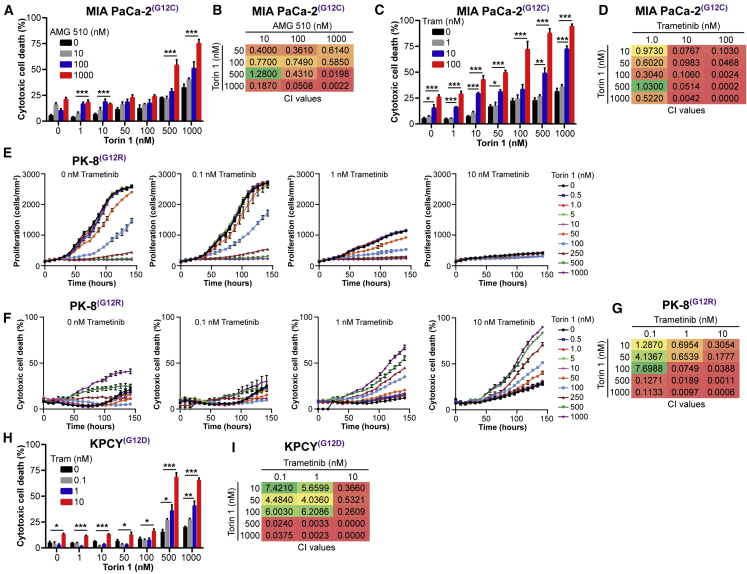
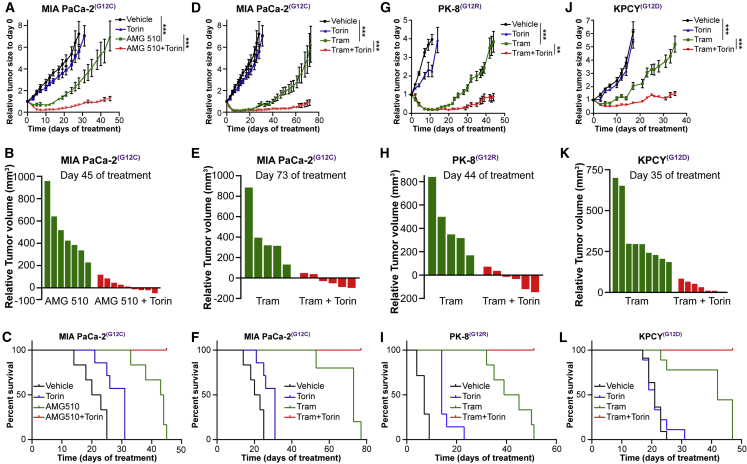
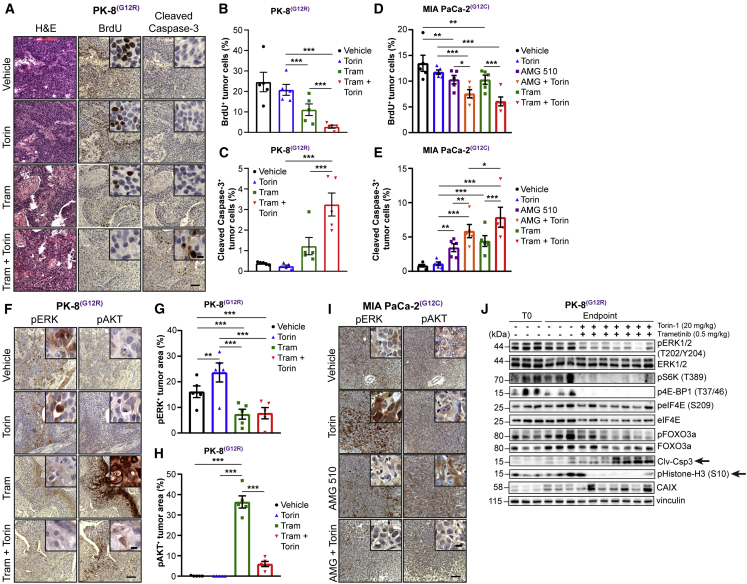
Activating KRAS mutations are found in over 90% of pancreatic ductal adenocarcinomas (PDACs), yet KRAS has remained a difficult target to inhibit pharmacologically. Here, we demonstrate, using several human and mouse models of PDACs, rapid acquisition of tumor resistance in response to targeting KRAS or MEK, associated with integrin-linked kinase (ILK)-mediated increased phosphorylation of the mTORC2 component Rictor, and AKT. Although inhibition of mTORC1/2 results in a compensatory increase in ERK phosphorylation, combinatorial treatment of PDAC cells with either KRAS (G12C) or MEK inhibitors, together with mTORC1/2 inhibitors, results in synergistic cytotoxicity and cell death reflected by inhibition of pERK and pRictor/pAKT and of downstream regulators of protein synthesis and cell survival. Relative to single agents alone, this combination leads to durable inhibition of tumor growth and metastatic progression in vivo and increased survival. We have identified an effective combinatorial treatment strategy using clinically viable inhibitors, which can be applied to PDAC tumors with different KRAS mutations.
Keywords: AMG 510; KRAS; PDAC; acquired resistance; cellular toxicity; protein translation; signal transduction; tumor regression.
© 2020 The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- Siegel R.L., Miller K.D., Jemal A. Cancer statistics, 2019. CA Cancer J. Clin. 2019;69:7–34. - PubMed
-
- Kleeff J., Korc M., Apte M., La Vecchia C., Johnson C.D., Biankin A.V., Neale R.E., Tempero M., Tuveson D.A., Hruban R.H., Neoptolemos J.P. Pancreatic cancer. Nat. Rev. Dis. Primers. 2016;2:16022. - PubMed
-
- Ducreux M., Seufferlein T., Van Laethem J.-L., Laurent-Puig P., Smolenschi C., Malka D., Boige V., Hollebecque A., Conroy T. Systemic treatment of pancreatic cancer revisited. Semin. Oncol. 2019;46:28–38. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
