

Cystathionine β-synthase deficiency in the E-HOD registry-part I: pyridoxine responsiveness as a determinant of biochemical and clinical phenotype at diagnosis
- PMID: 33295057
- PMCID: PMC8247016
- DOI: 10.1002/jimd.12338
Cystathionine β-synthase deficiency in the E-HOD registry-part I: pyridoxine responsiveness as a determinant of biochemical and clinical phenotype at diagnosis
Abstract
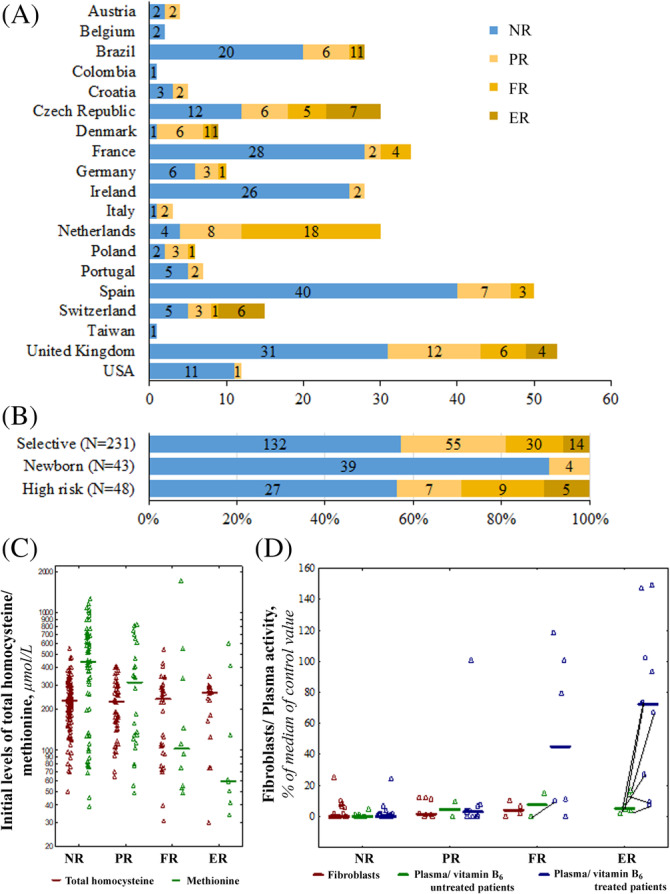
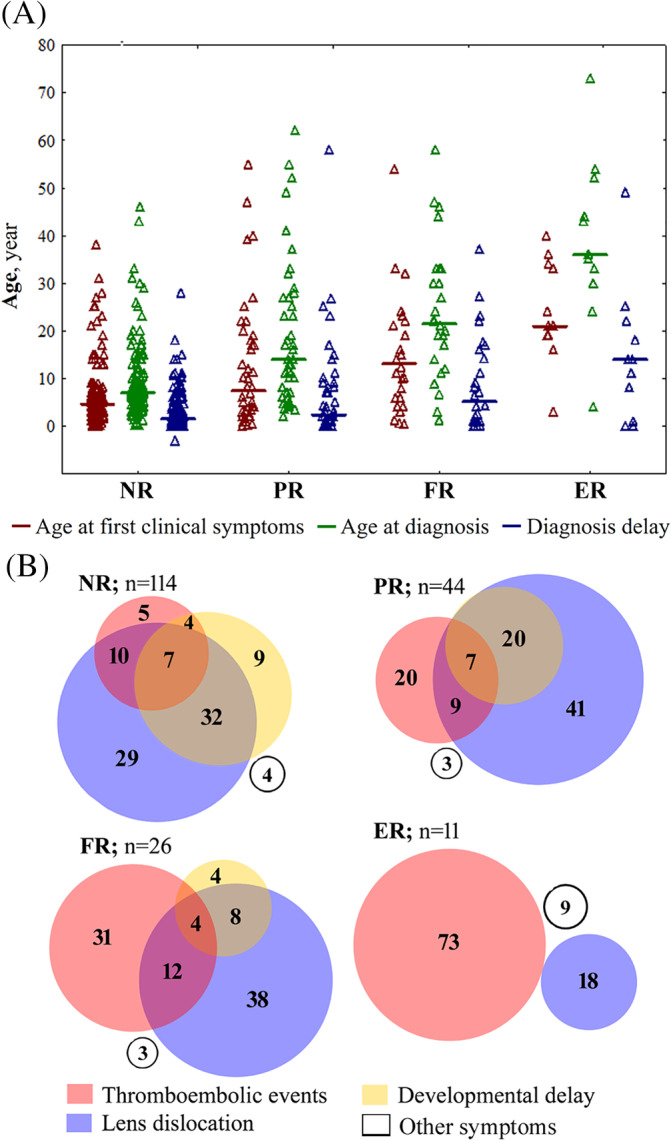
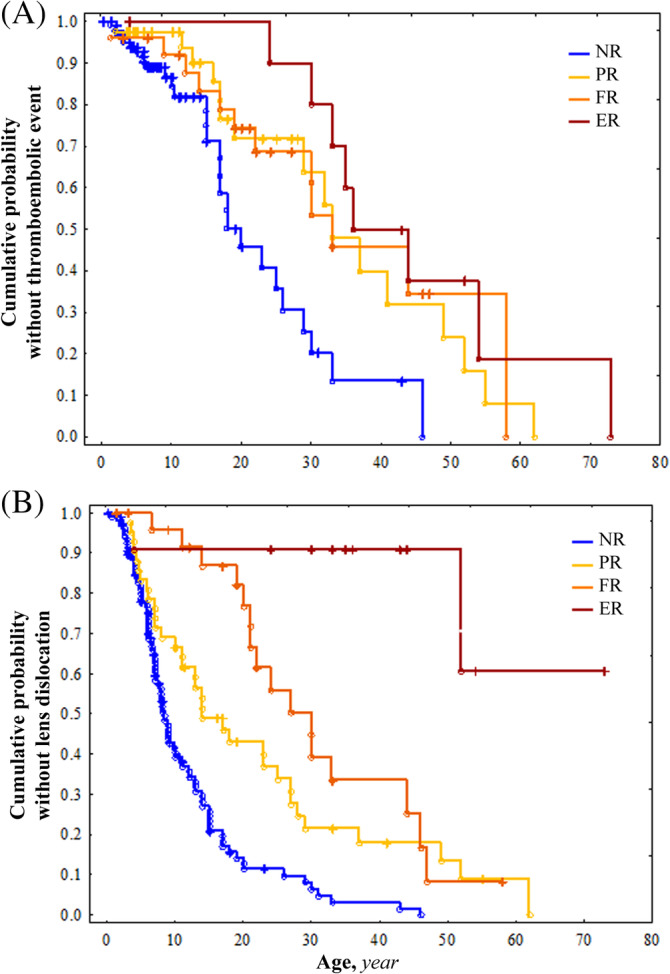
Cystathionine β-synthase (CBS) deficiency has a wide clinical spectrum, ranging from neurodevelopmental problems, lens dislocation and marfanoid features in early childhood to adult onset disease with predominantly thromboembolic complications. We have analysed clinical and laboratory data at the time of diagnosis in 328 patients with CBS deficiency from the E-HOD (European network and registry for Homocystinurias and methylation Defects) registry. We developed comprehensive criteria to classify patients into four groups of pyridoxine responsivity: non-responders (NR), partial, full and extreme responders (PR, FR and ER, respectively). All groups showed overlapping concentrations of plasma total homocysteine while pyridoxine responsiveness inversely correlated with plasma/serum methionine concentrations. The FR and ER groups had a later age of onset and diagnosis and a longer diagnostic delay than NR and PR patients. Lens dislocation was common in all groups except ER but the age of dislocation increased with increasing responsiveness. Developmental delay was commonest in the NR group while no ER patient had cognitive impairment. Thromboembolism was the commonest presenting feature in ER patients, whereas it was least likely at presentation in the NR group. This probably is due to the differences in ages at presentation: all groups had a similar number of thromboembolic events per 1000 patient-years. Clinical severity of CBS deficiency depends on the degree of pyridoxine responsiveness. Therefore, a standardised pyridoxine-responsiveness test in newly diagnosed patients and a critical review of previous assessments is indispensable to ensure adequate therapy and to prevent or reduce long-term complications.
Keywords: developmental delay; homocystinuria; methionine; natural history; patient registry; thromboembolism.
© 2020 The Authors. Journal of Inherited Metabolic Disease published by John Wiley & Sons Ltd on behalf of SSIEM.
Conflict of interest statement
Viktor Kožich, Jitka Sokolová and Jakub Krijt declare that Charles University‐First Faculty of Medicine received a partial reimbursement for preclinical testing of OT‐58 from Orphan Technologies. Andrew A.M. Morris received honoraria for lectures from Recordati & Nutricia and Advisory Board fees from Nutricia; he is an Expert Witness for a relevant medicolegal case. Stefan Kölker received funding from Recordati Rare Diseases for postmarketing authorization studies on Cystadane (betaine anhydrous). Martina Huemer has received honoraria for a lecture from Aeglea and research grant from Nutricia Metabolics. Carlo Dionisi‐Vici received a research fund and reimbursement for attending a symposium from Medifood, honoraria from Recordati‐Orphan Europe and Nutricia, and consulting fee from Nutricia. Markéta Pavlíková, Florian Gleich, Henk Blom and Matthias M.R. Baumgartner declare that they have no conflict of interest.
Figures
Similar articles
-
Cystathionine β-Synthase Deficiency in the E-HOD Registry-Part II: Dietary and Pharmacological Treatment.J Inherit Metab Dis. 2025 Jan;48(1):e12844. doi: 10.1002/jimd.12844. J Inherit Metab Dis. 2025. PMID: 40095936 Free PMC article.
-
High prevalence of cerebral venous sinus thrombosis (CVST) as presentation of cystathionine beta-synthase deficiency in childhood: molecular and clinical findings of Turkish probands.Gene. 2014 Jan 25;534(2):197-203. doi: 10.1016/j.gene.2013.10.060. Epub 2013 Nov 6. Gene. 2014. PMID: 24211323
-
Homocystinuria due to cystathionine beta-synthase deficiency--the effects of betaine treatment in pyridoxine-responsive patients.Metabolism. 1985 Dec;34(12):1115-21. doi: 10.1016/0026-0495(85)90156-8. Metabolism. 1985. PMID: 3934499
-
Recent advances in the mechanism of pyridoxine-responsive disorders.J Inherit Metab Dis. 1985;8 Suppl 1:76-83. doi: 10.1007/BF01800664. J Inherit Metab Dis. 1985. PMID: 3930845 Review.
-
Komrower Memorial Lecture 2023. Molecular basis of phenotype expression in homocystinuria: Where are we 30 years later?J Inherit Metab Dis. 2024 Sep;47(5):841-859. doi: 10.1002/jimd.12767. Epub 2024 Jun 14. J Inherit Metab Dis. 2024. PMID: 38873792 Review.
Cited by
-
Classical homocystinuria, is it safe to exercise?Mol Genet Metab Rep. 2021 Mar 26;27:100746. doi: 10.1016/j.ymgmr.2021.100746. eCollection 2021 Jun. Mol Genet Metab Rep. 2021. PMID: 33868930 Free PMC article.
-
Rare Disease Registries Are Key to Evidence-Based Personalized Medicine: Highlighting the European Experience.Front Endocrinol (Lausanne). 2022 Mar 4;13:832063. doi: 10.3389/fendo.2022.832063. eCollection 2022. Front Endocrinol (Lausanne). 2022. PMID: 35317224 Free PMC article. Review.
-
Pathogenic Homocystinuria-Associated T236N Mutation Dramatically Alters the Biochemical Properties of Cystathionine Beta-Synthase Protein.Biomedicines. 2024 Apr 23;12(5):929. doi: 10.3390/biomedicines12050929. Biomedicines. 2024. PMID: 38790892 Free PMC article.
-
Cystathionine Beta-Synthase Deficiency: Three Consecutive Cases Detected in 40 Days by Newborn Screening in Emilia Romagna (Italy) and a Comprehensive Review of the Literature.Children (Basel). 2023 Feb 17;10(2):396. doi: 10.3390/children10020396. Children (Basel). 2023. PMID: 36832525 Free PMC article.
-
Examination of two different proteasome inhibitors in reactivating mutant human cystathionine β-synthase in mice.PLoS One. 2023 Jun 15;18(6):e0286550. doi: 10.1371/journal.pone.0286550. eCollection 2023. PLoS One. 2023. PMID: 37319242 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
