

Inhibition of macrophage proliferation dominates plaque regression in response to cholesterol lowering
- PMID: 33296022
- PMCID: PMC7725697
- DOI: 10.1007/s00395-020-00838-4
Inhibition of macrophage proliferation dominates plaque regression in response to cholesterol lowering
Abstract
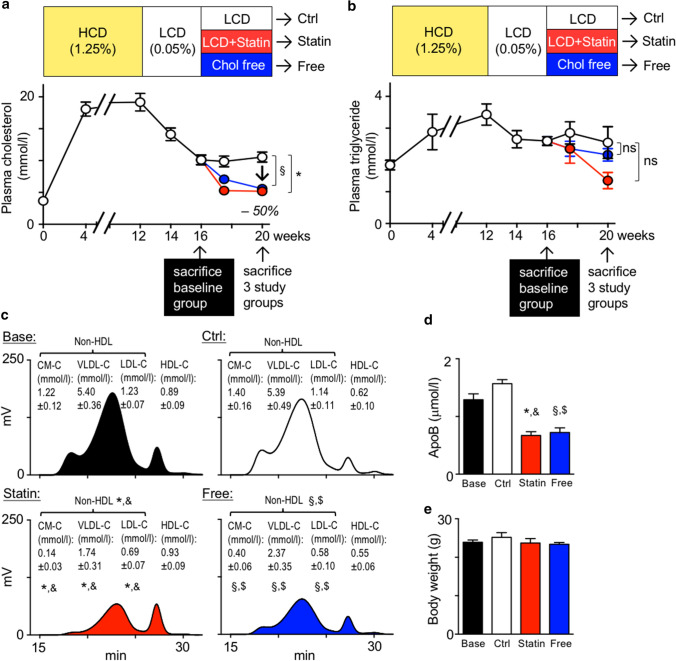
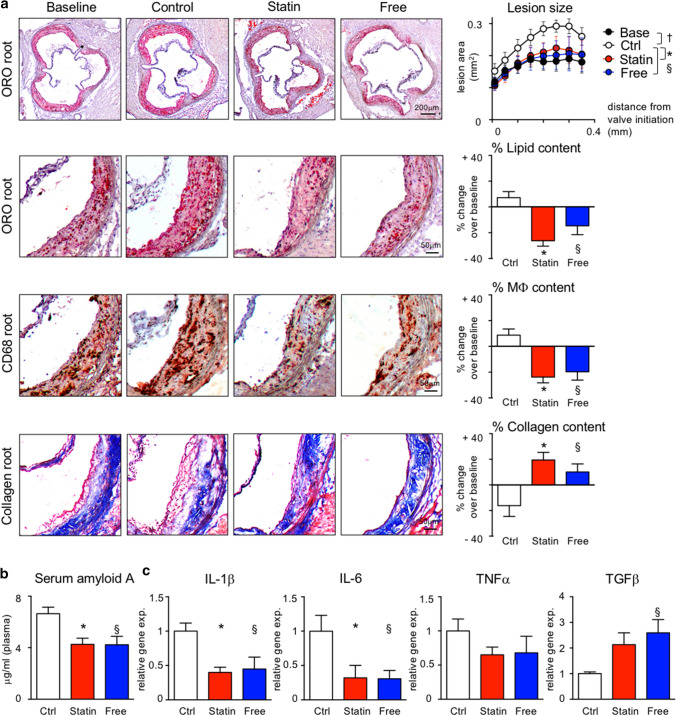
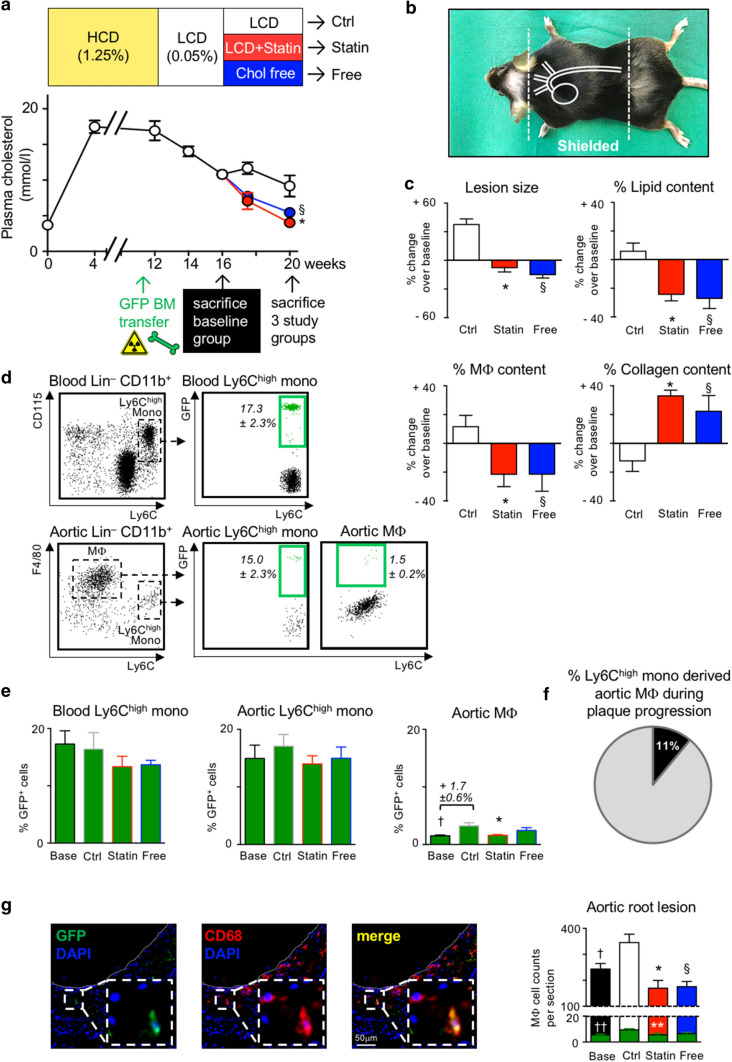
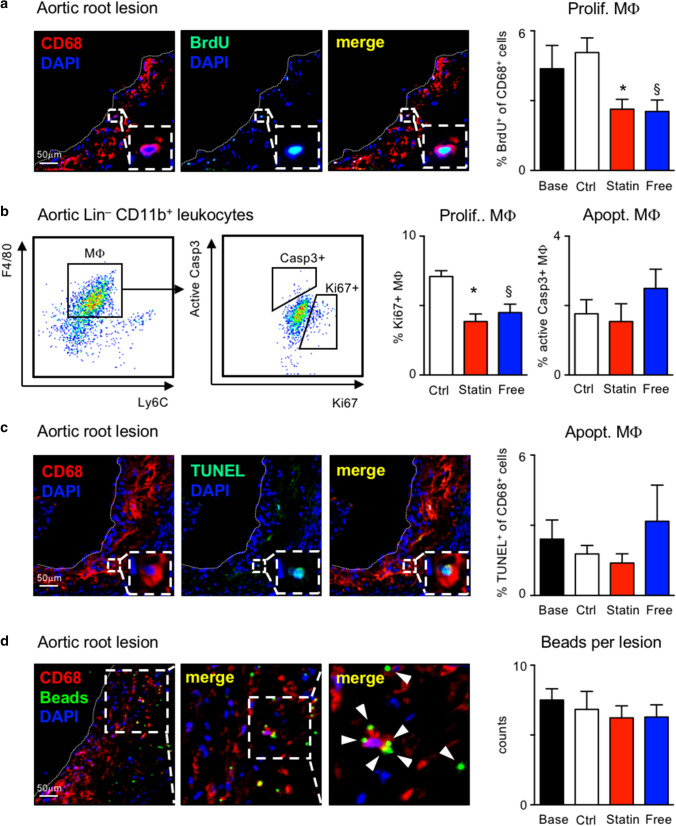
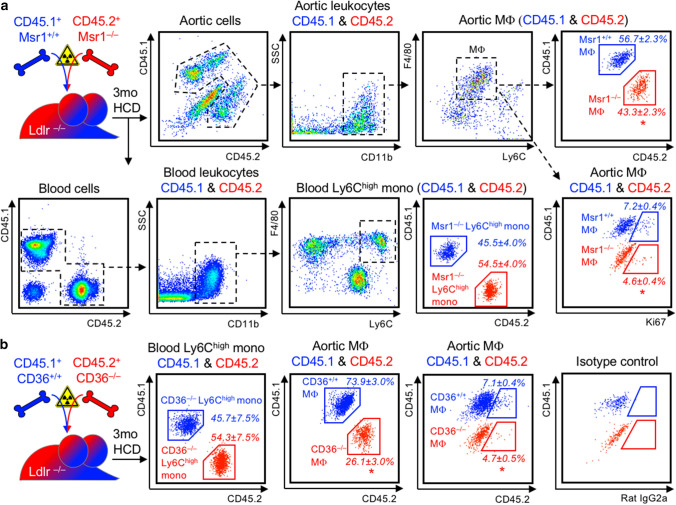
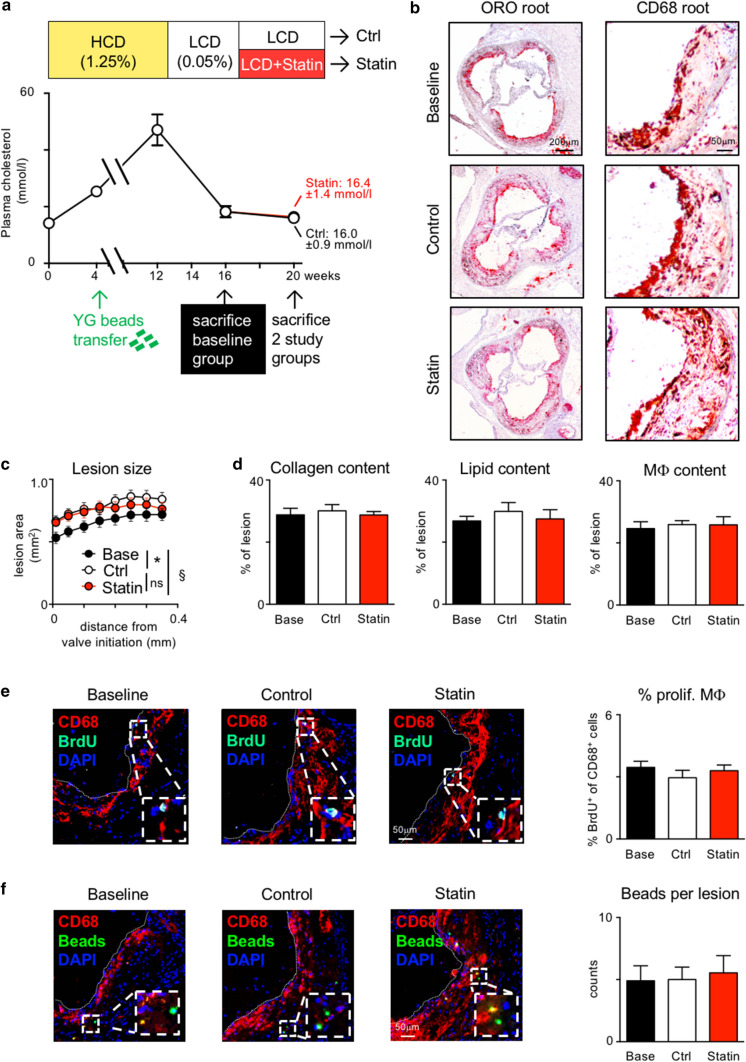
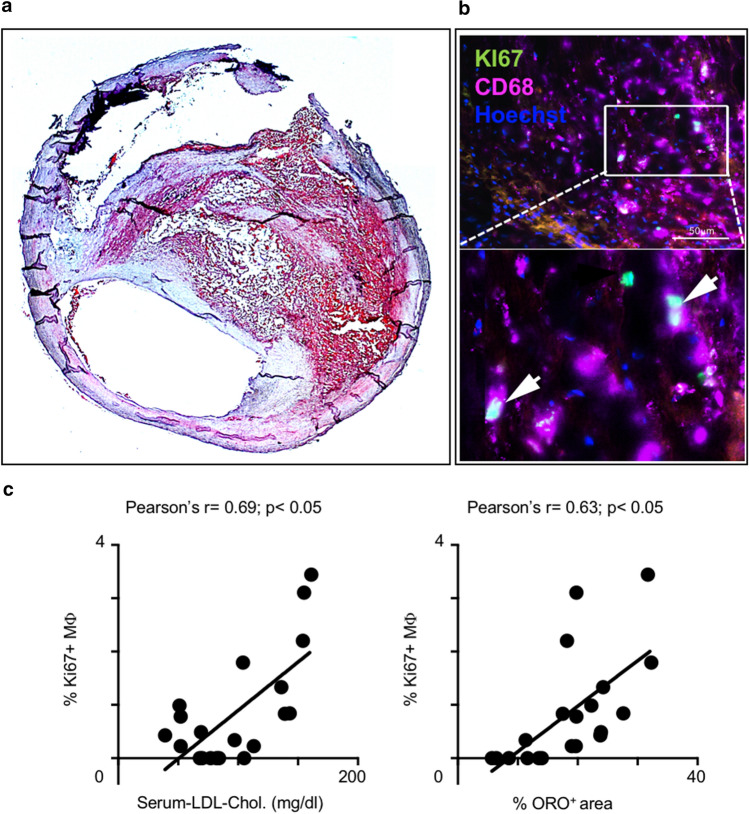
Statins induce plaque regression characterized by reduced macrophage content in humans, but the underlying mechanisms remain speculative. Studying the translational APOE*3-Leiden.CETP mouse model with a humanized lipoprotein metabolism, we find that systemic cholesterol lowering by oral atorvastatin or dietary restriction inhibits monocyte infiltration, and reverses macrophage accumulation in atherosclerotic plaques. Contrary to current believes, none of (1) reduced monocyte influx (studied by cell fate mapping in thorax-shielded irradiation bone marrow chimeras), (2) enhanced macrophage egress (studied by fluorescent bead labeling and transfer), or (3) atorvastatin accumulation in murine or human plaque (assessed by mass spectrometry) could adequately account for the observed loss in macrophage content in plaques that undergo phenotypic regression. Instead, suppression of local proliferation of macrophages dominates phenotypic plaque regression in response to cholesterol lowering: the lower the levels of serum LDL-cholesterol and lipid contents in murine aortic and human carotid artery plaques, the lower the rates of in situ macrophage proliferation. Our study identifies macrophage proliferation as the predominant turnover determinant and an attractive target for inducing plaque regression.
Keywords: Atherosclerosis; Macrophage; Plaque regression; Proliferation.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
Rosuvastatin Reduces Aortic Sinus and Coronary Artery Atherosclerosis in SR-B1 (Scavenger Receptor Class B Type 1)/ApoE (Apolipoprotein E) Double Knockout Mice Independently of Plasma Cholesterol Lowering.Arterioscler Thromb Vasc Biol. 2018 Jan;38(1):26-39. doi: 10.1161/ATVBAHA.117.305140. Epub 2017 Nov 21. Arterioscler Thromb Vasc Biol. 2018. PMID: 29162602 Free PMC article.
-
In vivo fluorescence-mediated tomography imaging demonstrates atorvastatin-mediated reduction of lesion macrophages in ApoE-/- mice.Anesthesiology. 2013 Jul;119(1):129-41. doi: 10.1097/ALN.0b013e318291c18b. Anesthesiology. 2013. PMID: 23559030
-
Apolipoprotein AI) Promotes Atherosclerosis Regression in Diabetic Mice by Suppressing Myelopoiesis and Plaque Inflammation.Circulation. 2019 Oct;140(14):1170-1184. doi: 10.1161/CIRCULATIONAHA.119.039476. Epub 2019 Sep 30. Circulation. 2019. PMID: 31567014 Free PMC article.
-
The phenomenon of atherosclerosis reversal and regression: Lessons from animal models.Exp Mol Pathol. 2017 Feb;102(1):138-145. doi: 10.1016/j.yexmp.2017.01.013. Epub 2017 Jan 17. Exp Mol Pathol. 2017. PMID: 28108216 Review.
-
Therapeutic strategies to deplete macrophages in atherosclerotic plaques.Br J Clin Pharmacol. 2012 Aug;74(2):246-63. doi: 10.1111/j.1365-2125.2012.04211.x. Br J Clin Pharmacol. 2012. PMID: 22309283 Free PMC article. Review.
Cited by
-
Myeloid cell-specific Irf5 deficiency stabilizes atherosclerotic plaques in Apoe-/- mice.Mol Metab. 2021 Nov;53:101250. doi: 10.1016/j.molmet.2021.101250. Epub 2021 May 12. Mol Metab. 2021. PMID: 33991749 Free PMC article.
-
Immune Homeostasis Maintenance Through Advanced Immune Therapeutics to Target Atherosclerosis.Methods Mol Biol. 2024;2782:25-37. doi: 10.1007/978-1-0716-3754-8_2. Methods Mol Biol. 2024. PMID: 38622390
-
Efferocytosis induces macrophage proliferation to help resolve tissue injury.Cell Metab. 2021 Dec 7;33(12):2445-2463.e8. doi: 10.1016/j.cmet.2021.10.015. Epub 2021 Nov 15. Cell Metab. 2021. PMID: 34784501 Free PMC article.
-
Roles of Macrophages in Atherogenesis.Front Pharmacol. 2021 Nov 26;12:785220. doi: 10.3389/fphar.2021.785220. eCollection 2021. Front Pharmacol. 2021. PMID: 34899348 Free PMC article. Review.
-
The Impact of Exercise on Immunity, Metabolism, and Atherosclerosis.Int J Mol Sci. 2023 Feb 8;24(4):3394. doi: 10.3390/ijms24043394. Int J Mol Sci. 2023. PMID: 36834808 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
