

DNA methyltransferase 3B deficiency unveils a new pathological mechanism of pulmonary hypertension
- PMID: 33298433
- PMCID: PMC7725449
- DOI: 10.1126/sciadv.aba2470
DNA methyltransferase 3B deficiency unveils a new pathological mechanism of pulmonary hypertension
Abstract
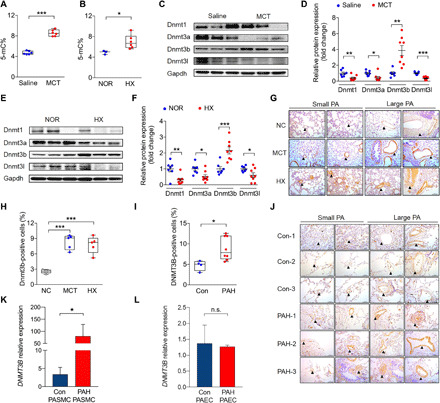
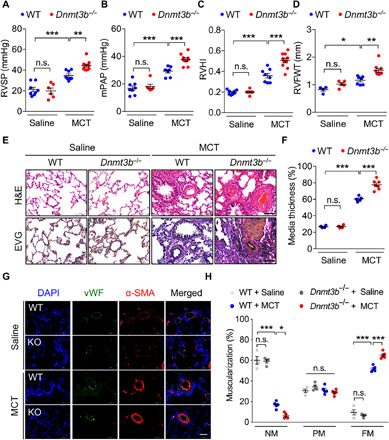
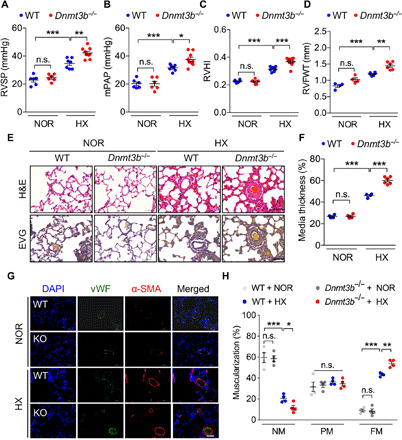
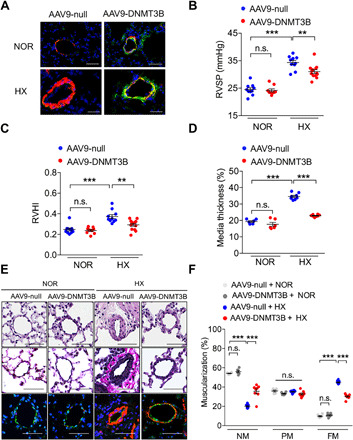
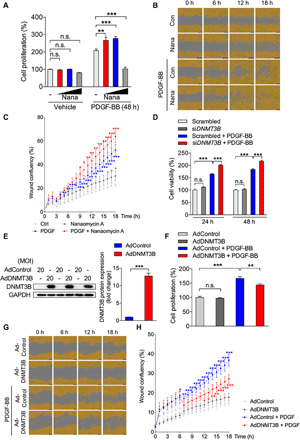
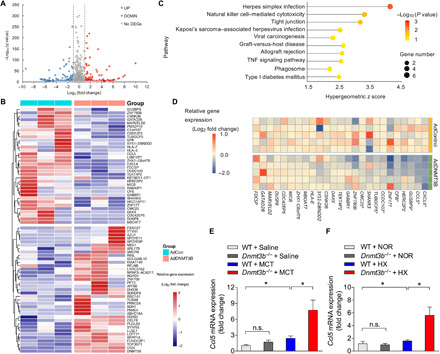
DNA methylation plays critical roles in vascular pathology of pulmonary hypertension (PH). The underlying mechanism, however, remains undetermined. Here, we demonstrate that global DNA methylation was elevated in the lungs of PH rat models after monocrotaline administration or hypobaric hypoxia exposure. We showed that DNA methyltransferase 3B (DNMT3B) was up-regulated in both PH patients and rodent models. Furthermore, Dnmt3b -/- rats exhibited more severe pulmonary vascular remodeling. Consistently, inhibition of DNMT3B promoted proliferation/migration of pulmonary artery smooth muscle cells (PASMCs) in response to platelet-derived growth factor-BB (PDGF-BB). In contrast, overexpressing DNMT3B in PASMCs attenuated PDGF-BB-induced proliferation/migration and ameliorated hypoxia-mediated PH and right ventricular hypertrophy in mice. We also showed that DNMT3B transcriptionally regulated inflammatory pathways. Our results reveal that DNMT3B is a previously undefined mediator in the pathogenesis of PH, which couples epigenetic regulations with vascular remodeling and represents a therapeutic target to tackle PH.
Copyright © 2020 The Authors, some rights reserved; exclusive licensee American Association for the Advancement of Science. No claim to original U.S. Government Works. Distributed under a Creative Commons Attribution NonCommercial License 4.0 (CC BY-NC).
Figures
References
-
- Farber H. W., Loscalzo J., Pulmonary arterial hypertension. N. Engl. J. Med. 351, 1655–1665 (2004). - PubMed
-
- Hoeper M. M., Bogaard H. J., Condliffe R., Frantz R., Khanna D., Kurzyna M., Langleben D., Manes A., Satoh T., Torres F., Wilkins M. R., Badesch D. B., Definitions and diagnosis of pulmonary hypertension. J. Am. Coll. Cardiol. 62 (suppl. 25), D42–D50 (2013). - PubMed
-
- Hopper R. K., Moonen J.-R. A. J., Diebold I., Cao A., Rhodes C. J., Tojais N. F., Hennigs J. K., Gu M., Wang L., Rabinovitch M., In pulmonary arterial hypertension, reduced BMPR2 promotes endothelial-to-mesenchymal transition via HMGA1 and its target slug. Circulation 133, 1783–1794 (2016). - PMC - PubMed
-
- Hwangbo C., Lee H.-W., Kang H., Ju H., Wiley D. S., Papangeli I., Han J., Kim J.-D., Dunworth W. P., Hu X., Lee S., El-Hely O., Sofer A., Pak B., Peterson L., Comhair S., Hwang E. M., Park J.-Y., Thomas J.-L., Bautch V. L., Erzurum S. C., Chun H. J., Jin S.-W., Modulation of endothelial bone morphogenetic protein receptor type 2 activity by vascular endothelial growth factor receptor 3 in pulmonary arterial hypertension. Circulation 135, 2288–2298 (2017). - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
