

Histone H1 loss drives lymphoma by disrupting 3D chromatin architecture
- PMID: 33299181
- PMCID: PMC7855728
- DOI: 10.1038/s41586-020-3017-y
Histone H1 loss drives lymphoma by disrupting 3D chromatin architecture
Abstract
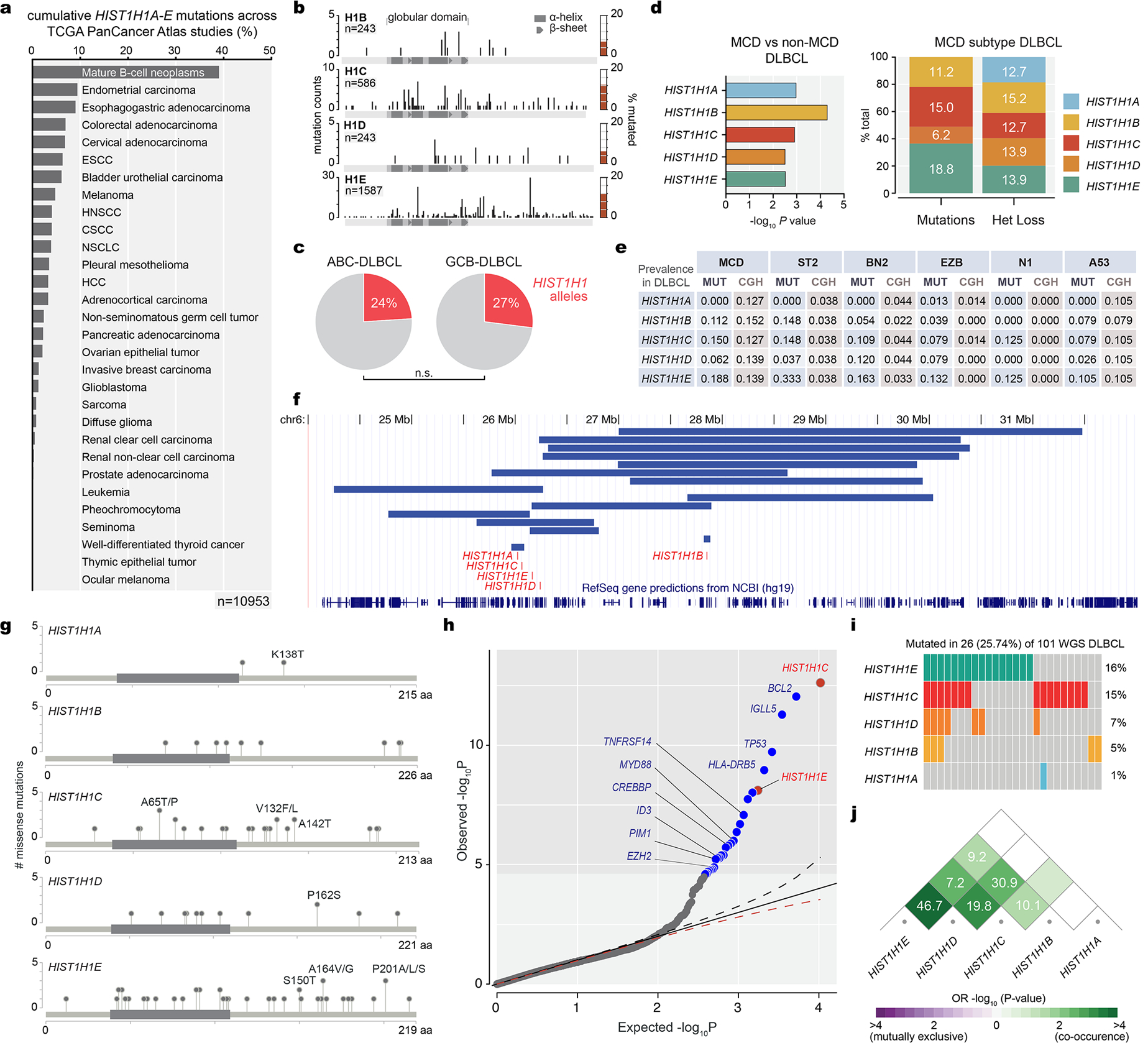
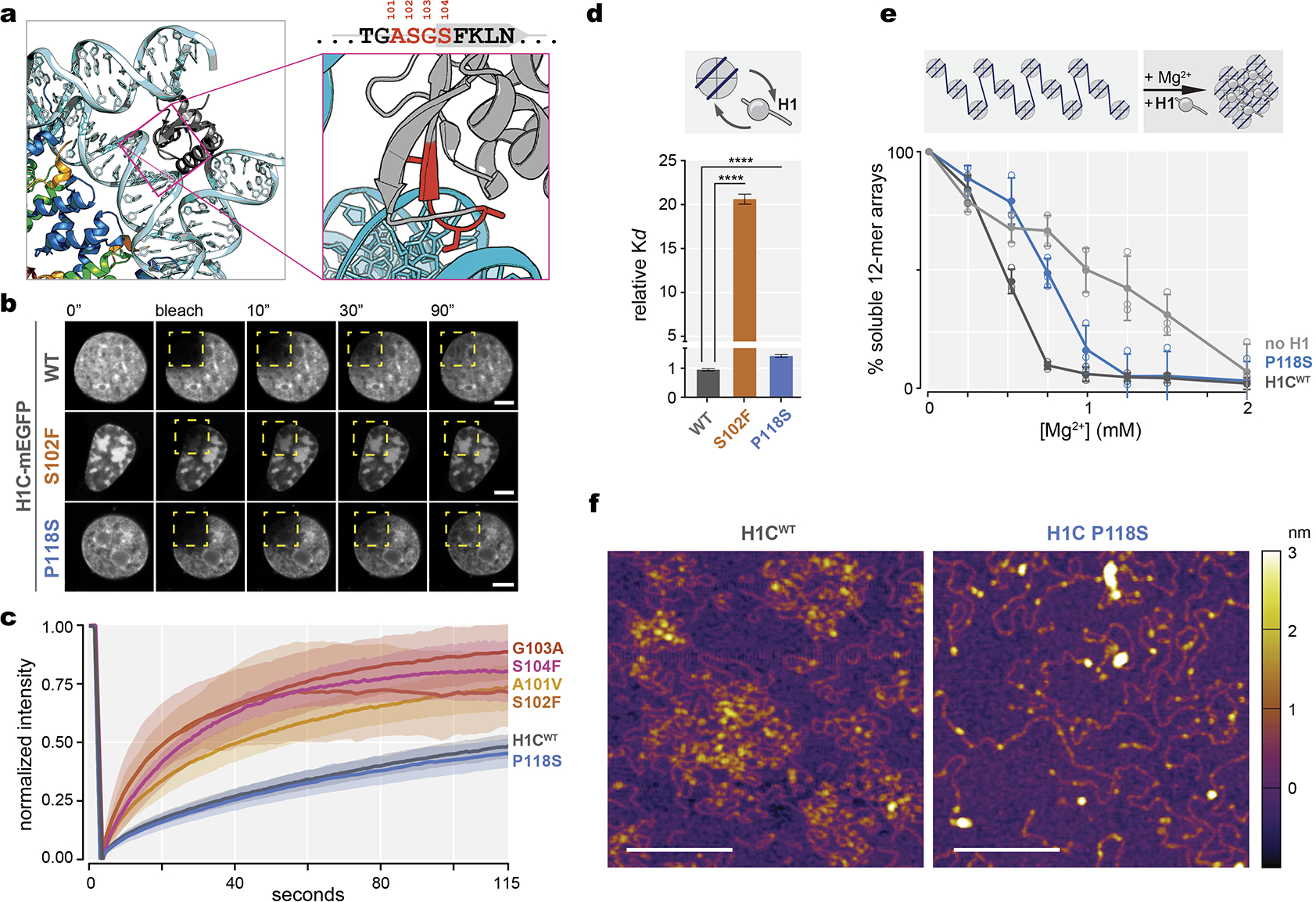
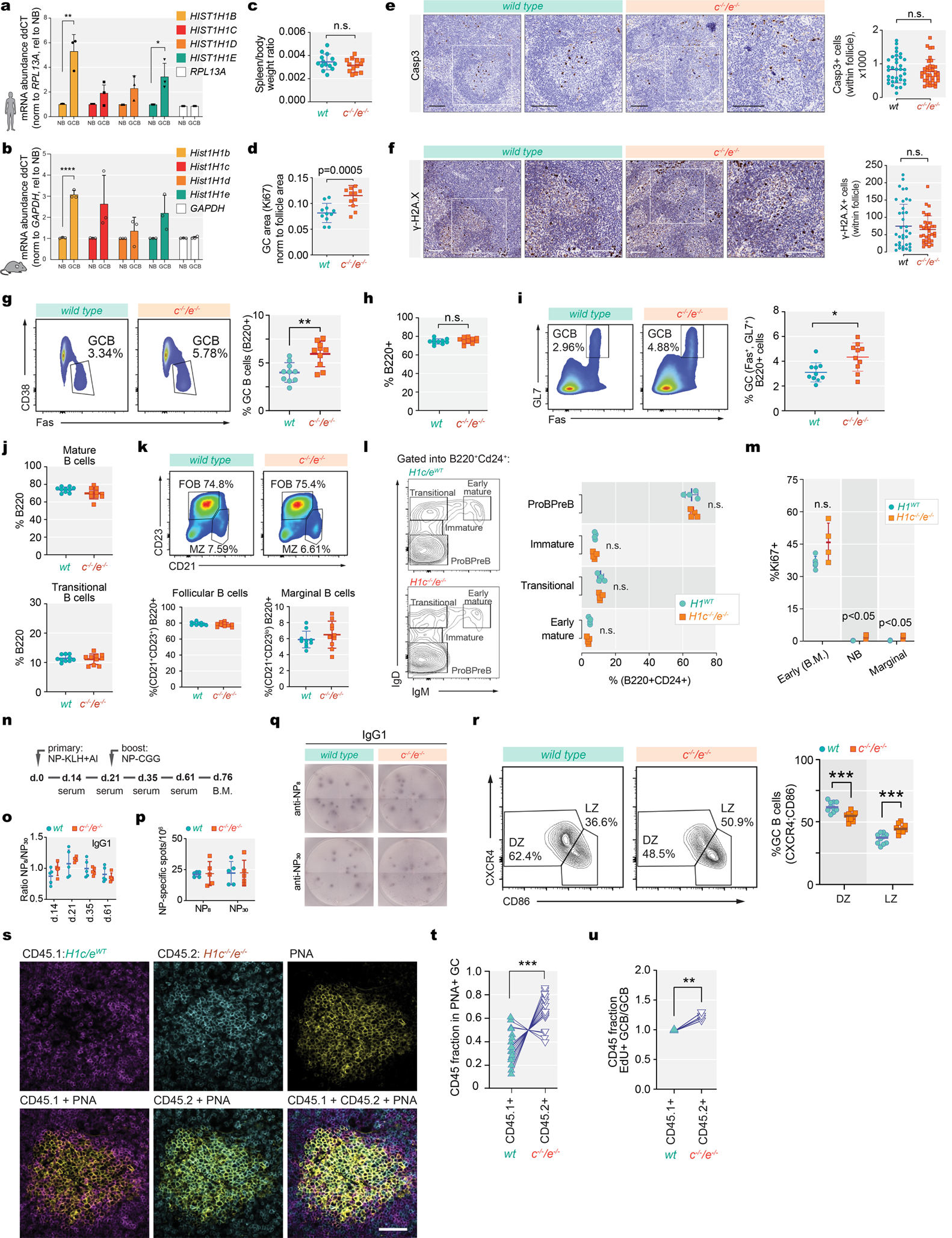
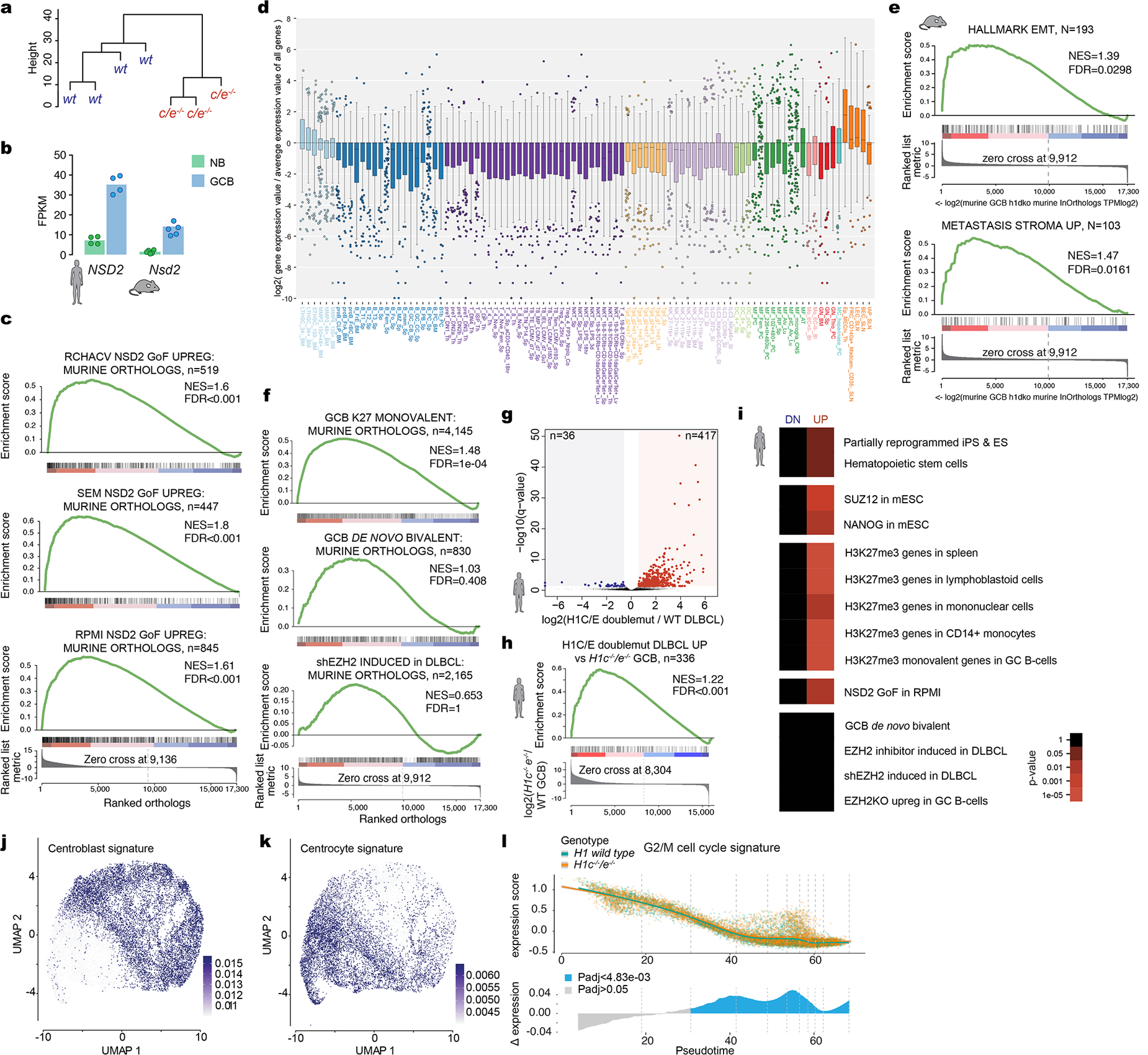
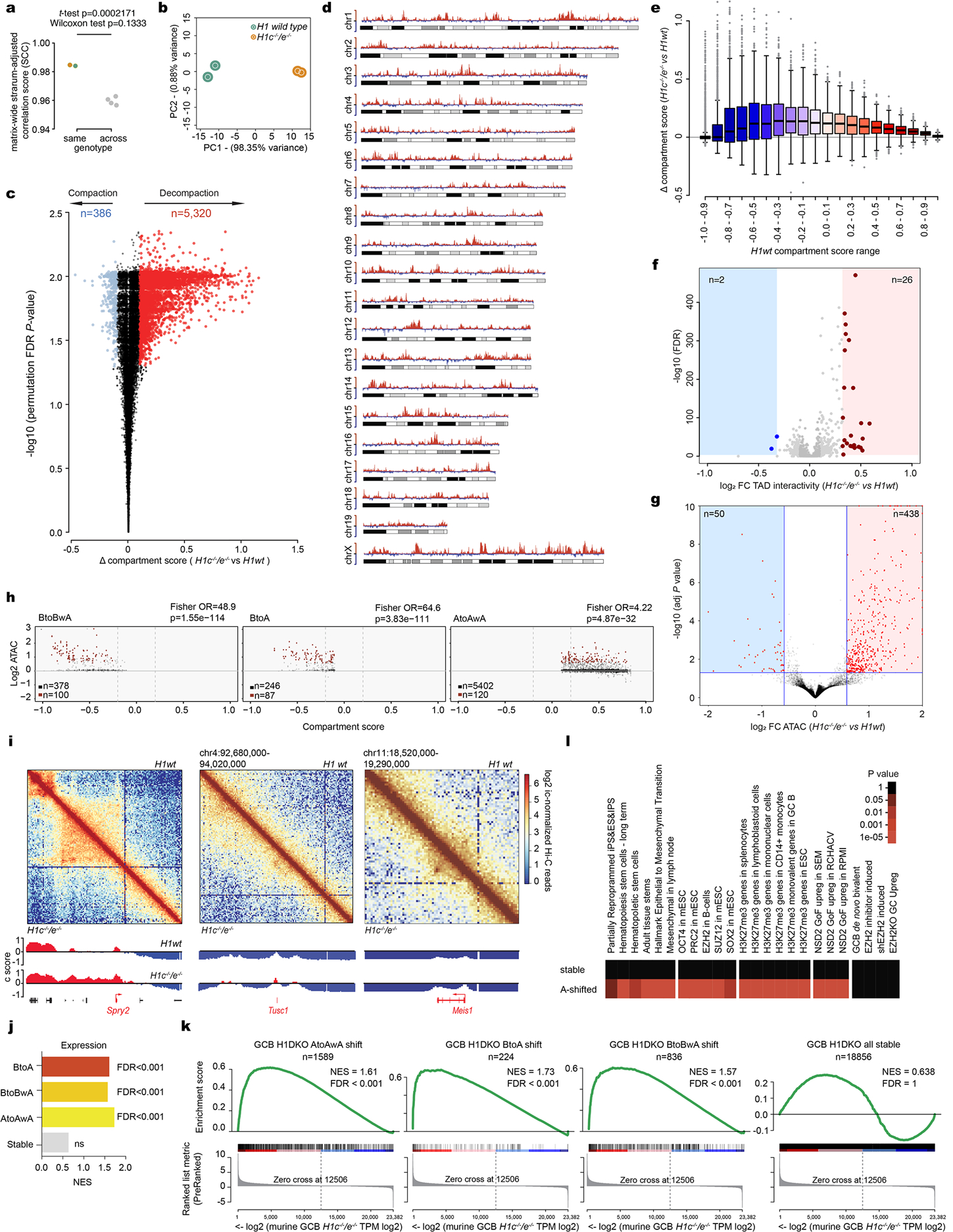
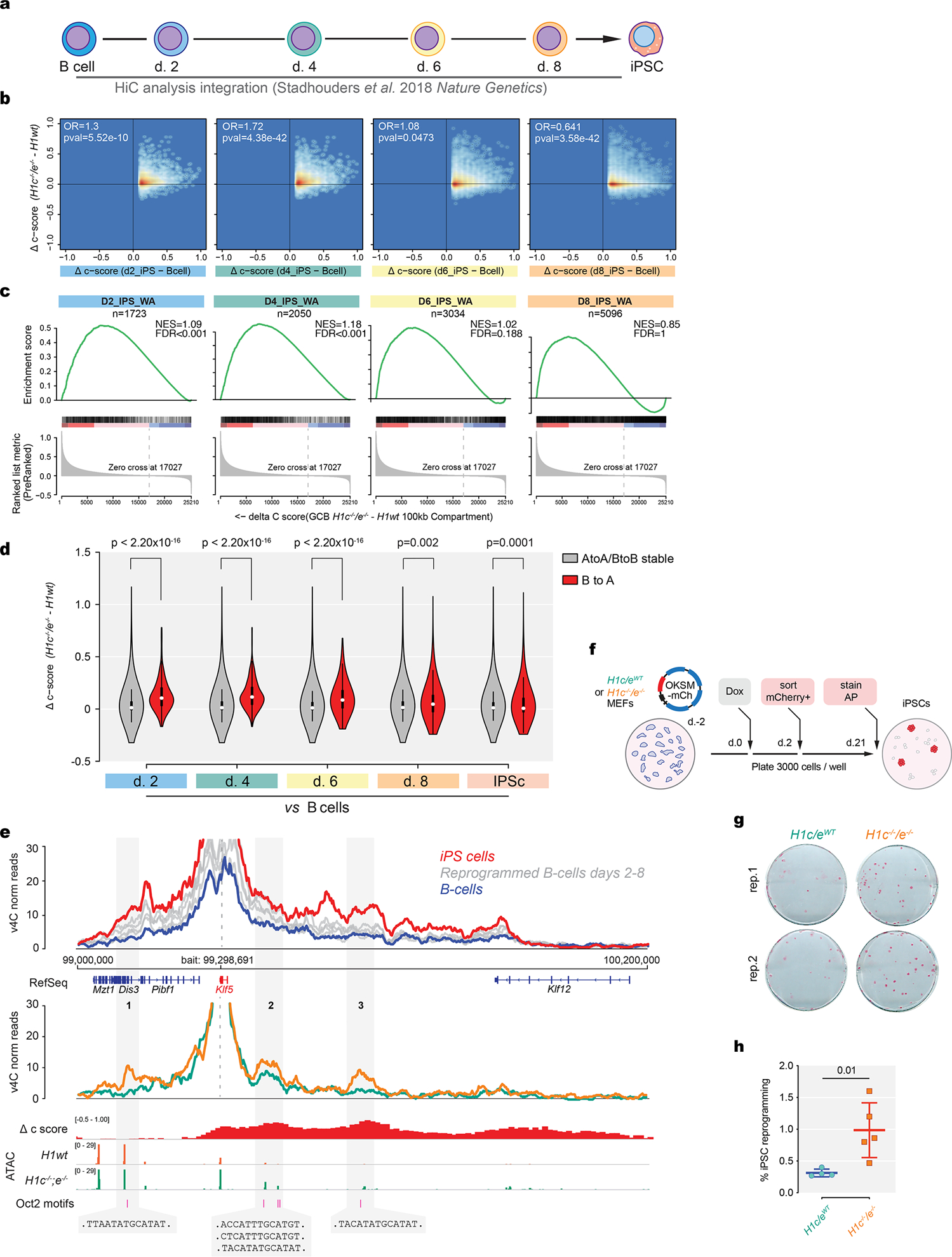
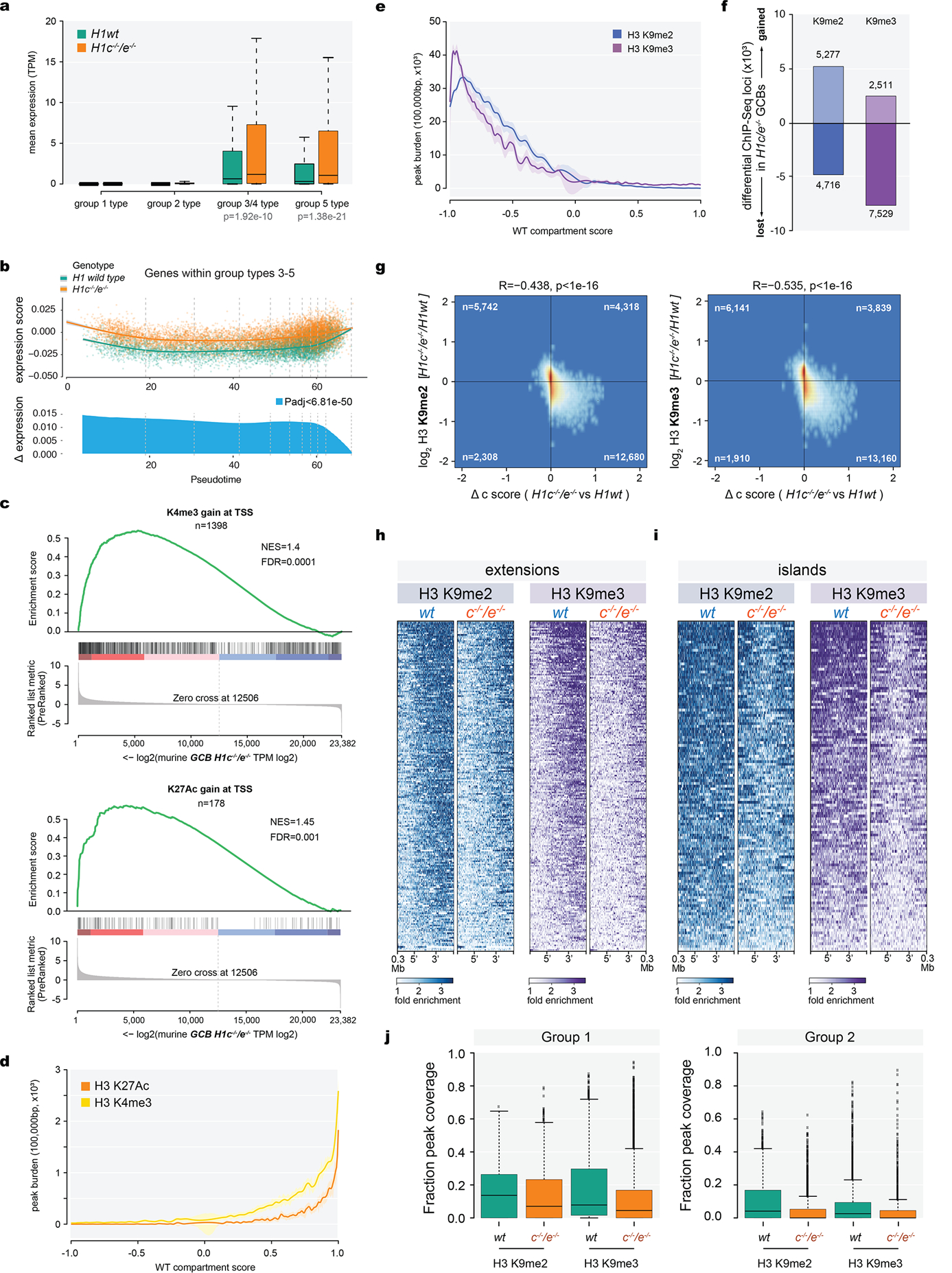
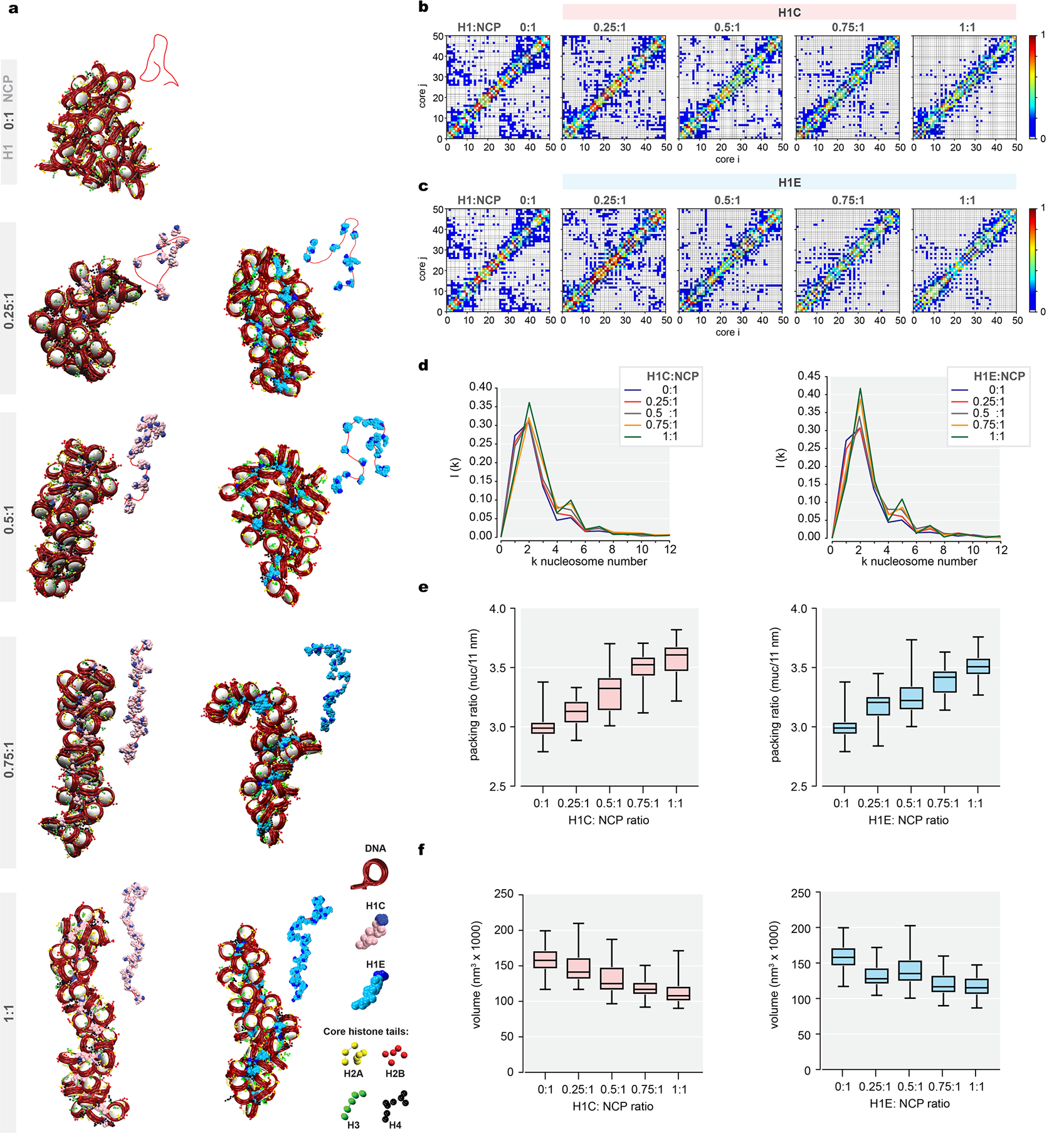
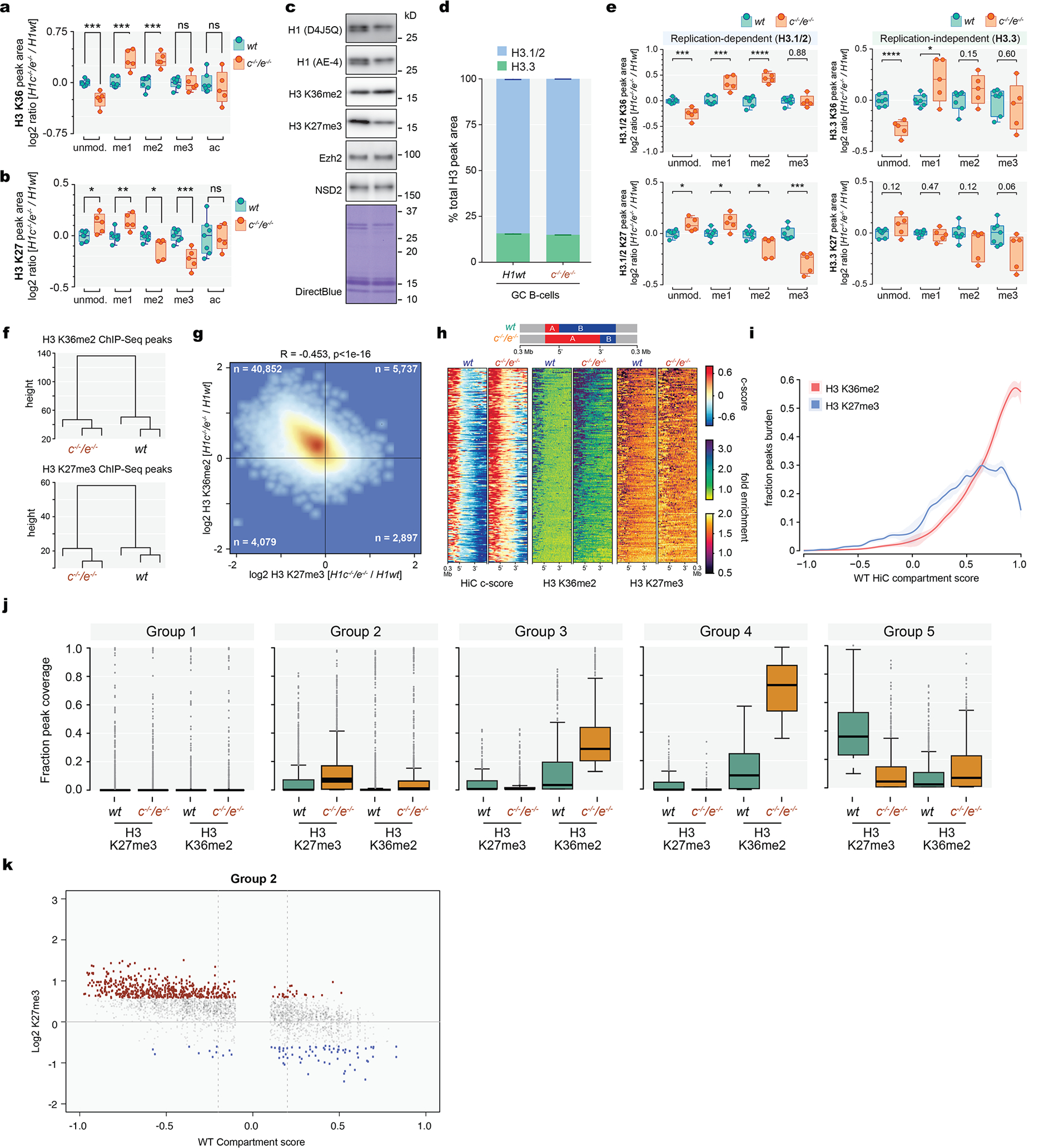
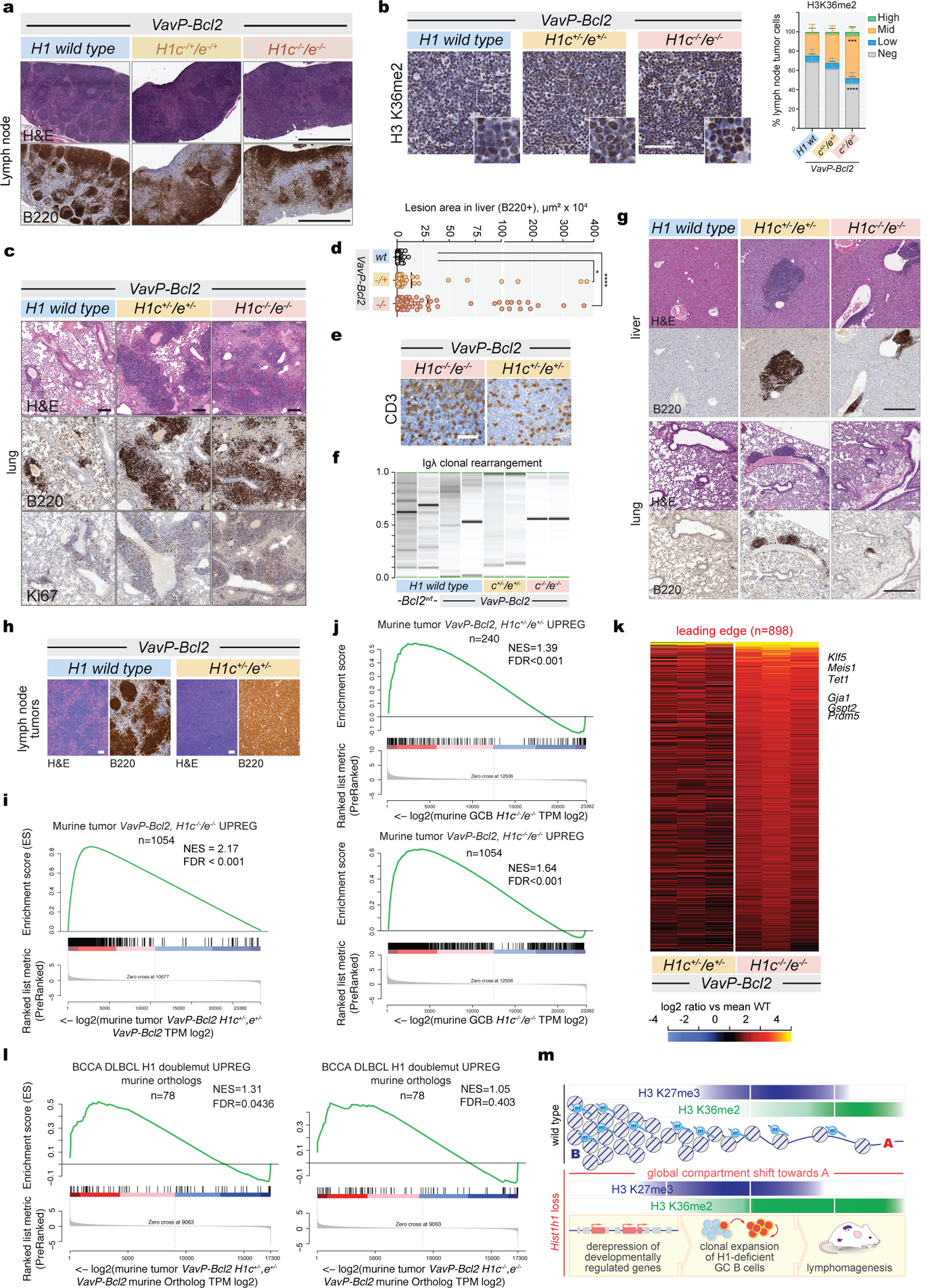
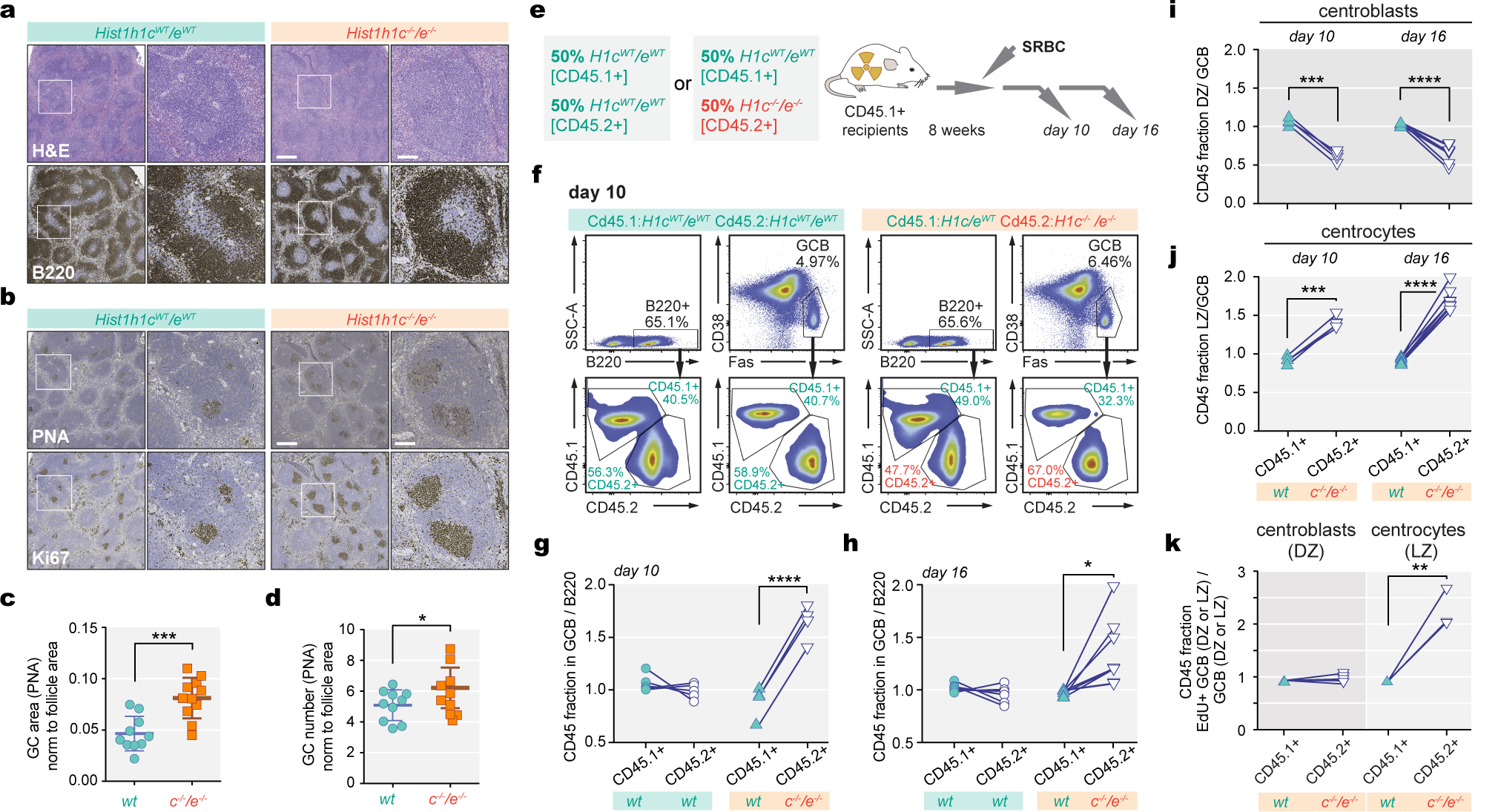
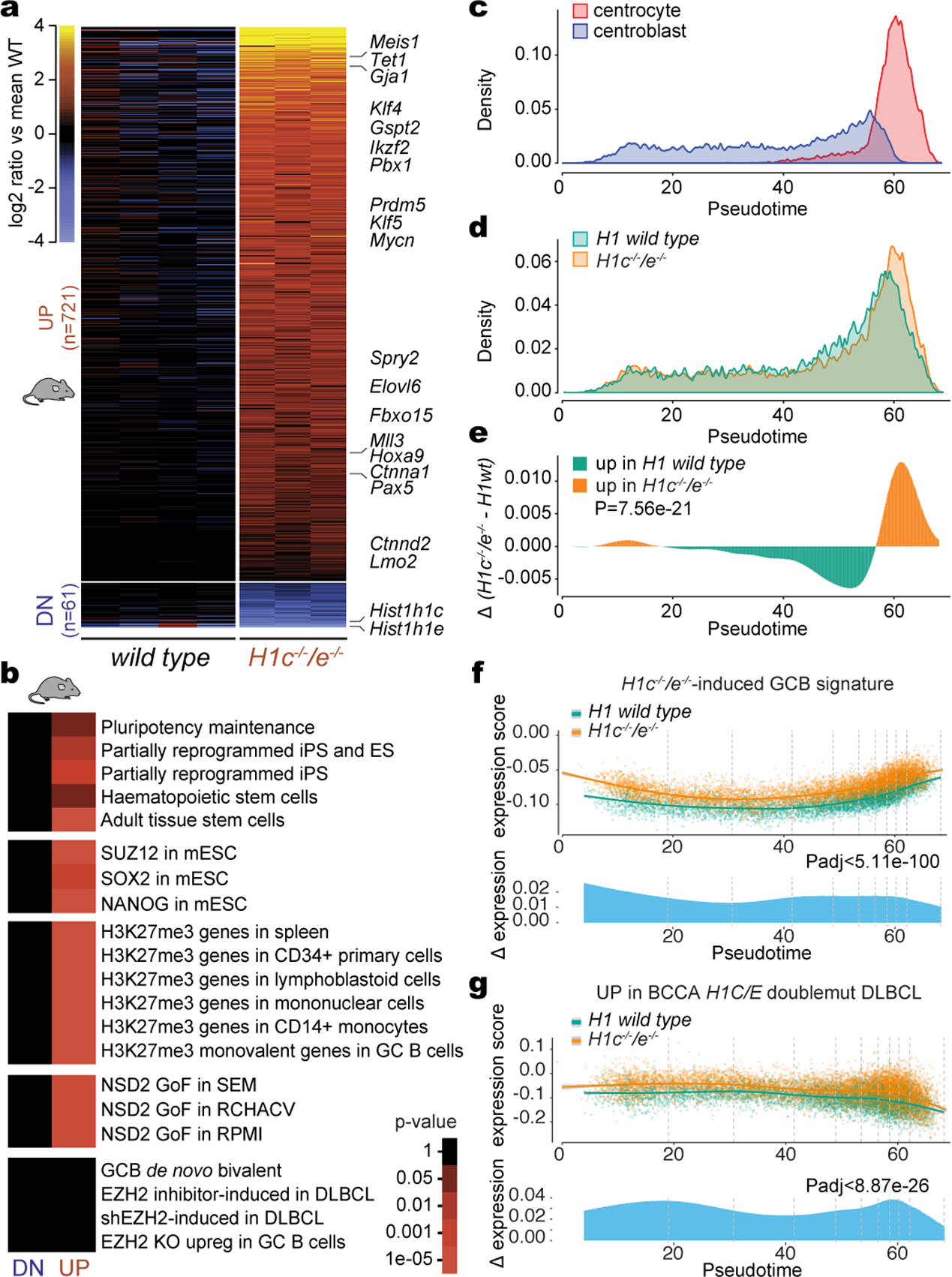
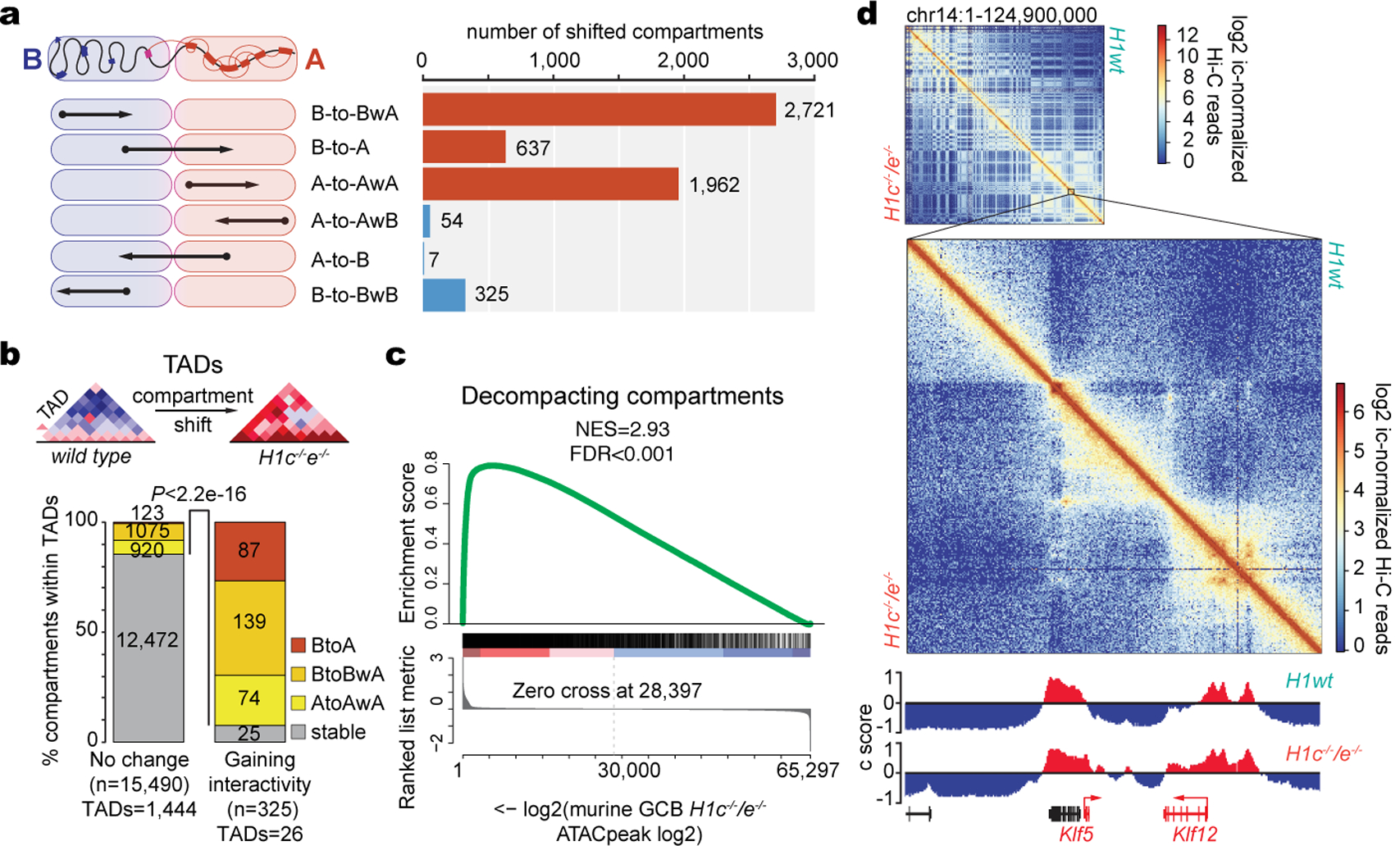
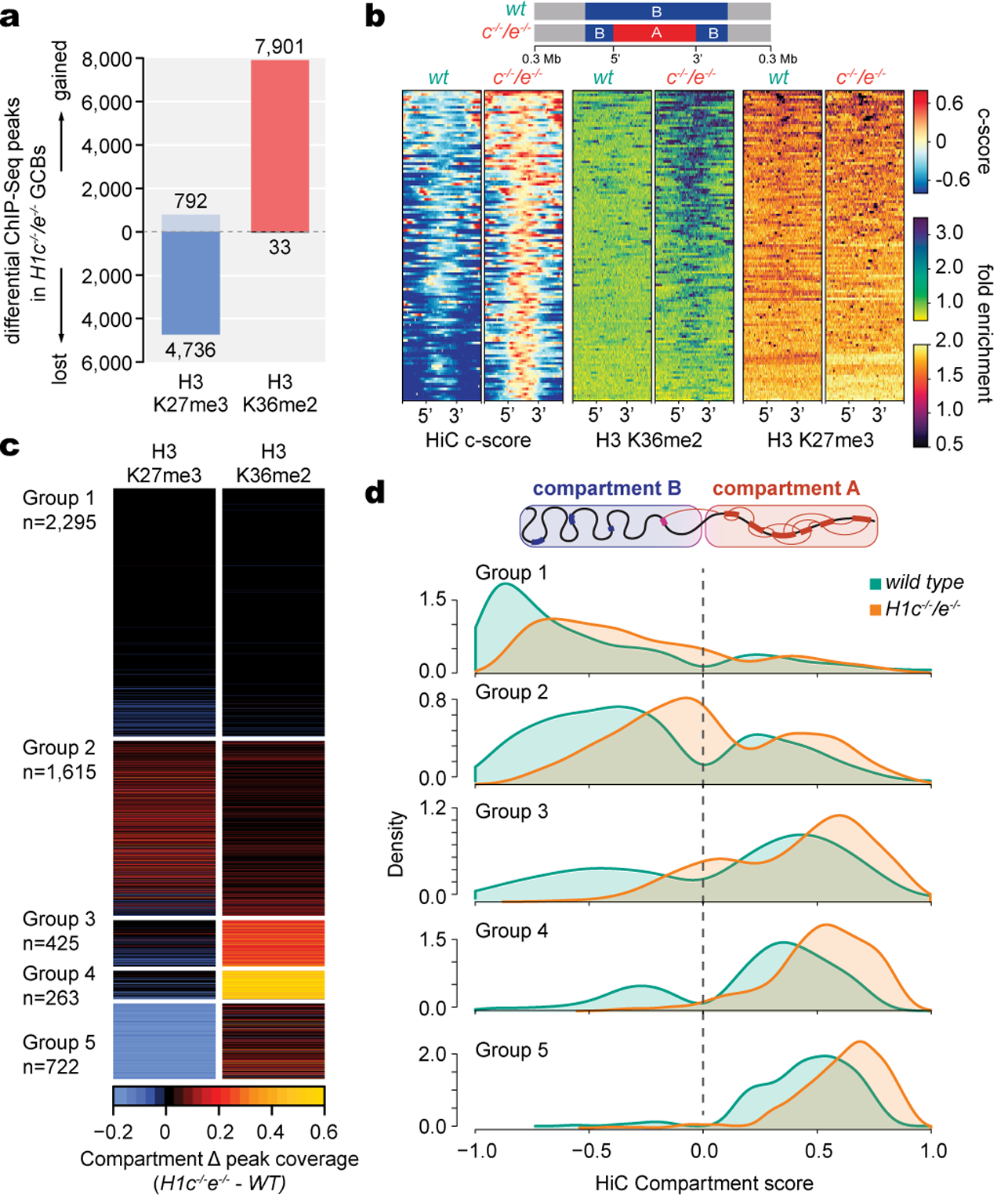
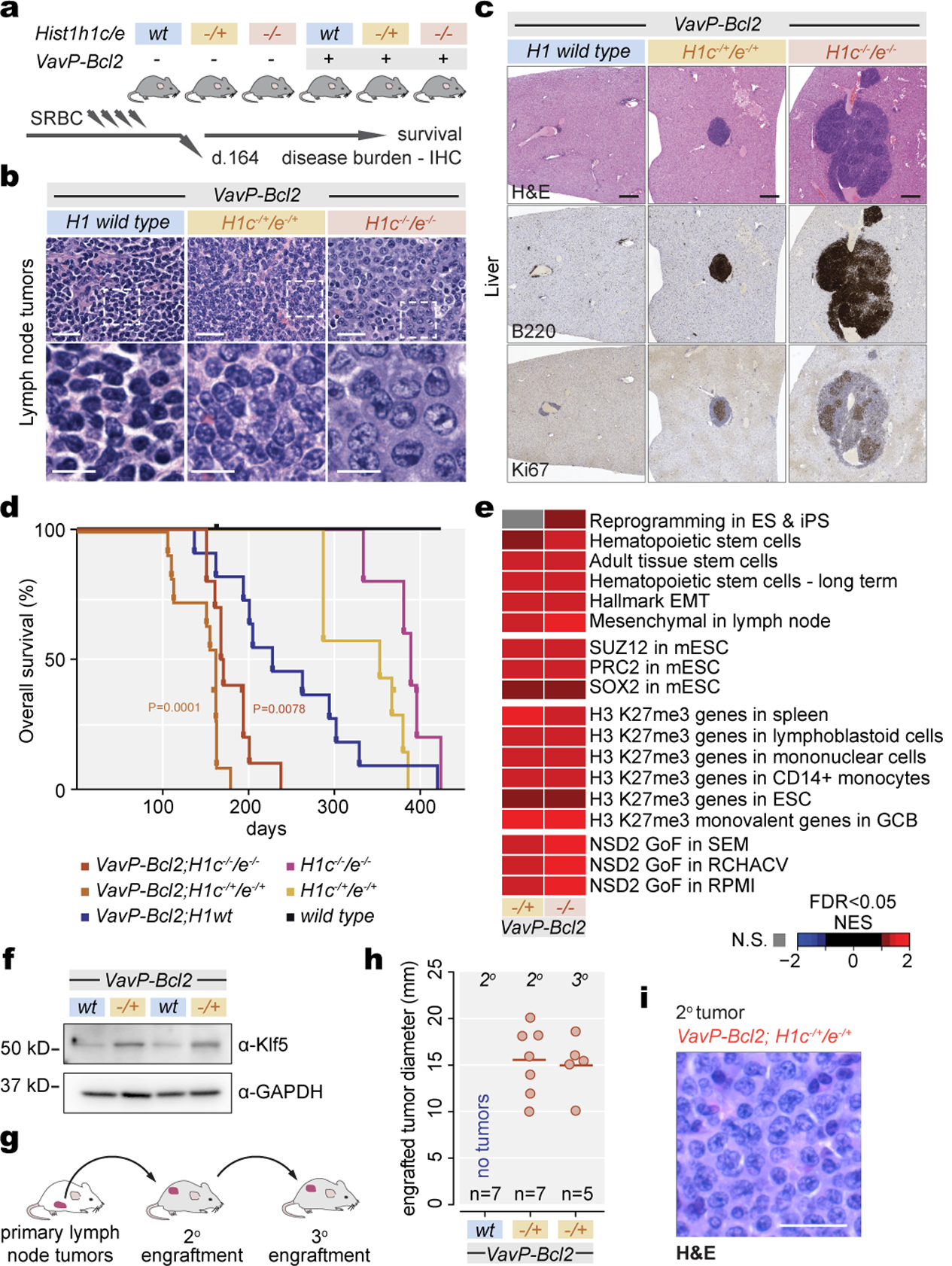
Linker histone H1 proteins bind to nucleosomes and facilitate chromatin compaction1, although their biological functions are poorly understood. Mutations in the genes that encode H1 isoforms B-E (H1B, H1C, H1D and H1E; also known as H1-5, H1-2, H1-3 and H1-4, respectively) are highly recurrent in B cell lymphomas, but the pathogenic relevance of these mutations to cancer and the mechanisms that are involved are unknown. Here we show that lymphoma-associated H1 alleles are genetic driver mutations in lymphomas. Disruption of H1 function results in a profound architectural remodelling of the genome, which is characterized by large-scale yet focal shifts of chromatin from a compacted to a relaxed state. This decompaction drives distinct changes in epigenetic states, primarily owing to a gain of histone H3 dimethylation at lysine 36 (H3K36me2) and/or loss of repressive H3 trimethylation at lysine 27 (H3K27me3). These changes unlock the expression of stem cell genes that are normally silenced during early development. In mice, loss of H1c and H1e (also known as H1f2 and H1f4, respectively) conferred germinal centre B cells with enhanced fitness and self-renewal properties, ultimately leading to aggressive lymphomas with an increased repopulating potential. Collectively, our data indicate that H1 proteins are normally required to sequester early developmental genes into architecturally inaccessible genomic compartments. We also establish H1 as a bona fide tumour suppressor and show that mutations in H1 drive malignant transformation primarily through three-dimensional genome reorganization, which leads to epigenetic reprogramming and derepression of developmentally silenced genes.
Conflict of interest statement
CONFLICT OF INTEREST
A.M. has research funding from Janssen Pharmaceuticals and Daiichi Sankyo, has consulted for Epizyme and Constellation and is on the advisory board for KDAC Pharma. NLK is a consultant for Thermo Fisher Scientific. C.D.A is a co-founder of Chroma Therapeutics and Constellation Pharmaceuticals, and a Scientific Advisory Board member of EpiCypher. EpiCypher is a commercial developer of the CUTANA® CUT&RUN platform. M.I. has received consultancy fees from Novartis Venture Fund outside of the scope of the work.
Figures
Comment in
-
Chromatin control by H1 histones.Nat Rev Genet. 2021 Feb;22(2):68-69. doi: 10.1038/s41576-020-00323-7. Nat Rev Genet. 2021. PMID: 33323999 No abstract available.
References
Publication types
MeSH terms
Substances
Grants and funding
- R01 CA252239/CA/NCI NIH HHS/United States
- P01 CA196539/CA/NCI NIH HHS/United States
- P41 GM108569/GM/NIGMS NIH HHS/United States
- P30 CA008748/CA/NCI NIH HHS/United States
- P30 CA060553/CA/NCI NIH HHS/United States
- R35 GM138386/GM/NIGMS NIH HHS/United States
- R01 CA195732/CA/NCI NIH HHS/United States
- S10 OD025194/OD/NIH HHS/United States
- R44 DE029633/DE/NIDCR NIH HHS/United States
- R35 CA220499/CA/NCI NIH HHS/United States
- P01 CA229086/CA/NCI NIH HHS/United States
- R35 GM122562/GM/NIGMS NIH HHS/United States
- R01 CA228528/CA/NCI NIH HHS/United States
- R01 GM116143/GM/NIGMS NIH HHS/United States
- R44 GM116584/GM/NIGMS NIH HHS/United States
- R01 CA234561/CA/NCI NIH HHS/United States
- R01 CA194547/CA/NCI NIH HHS/United States
- R01 GM147165/GM/NIGMS NIH HHS/United States
- U54 CA193419/CA/NCI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
