

H1 histones control the epigenetic landscape by local chromatin compaction
- PMID: 33299182
- PMCID: PMC8110206
- DOI: 10.1038/s41586-020-3032-z
H1 histones control the epigenetic landscape by local chromatin compaction
Abstract
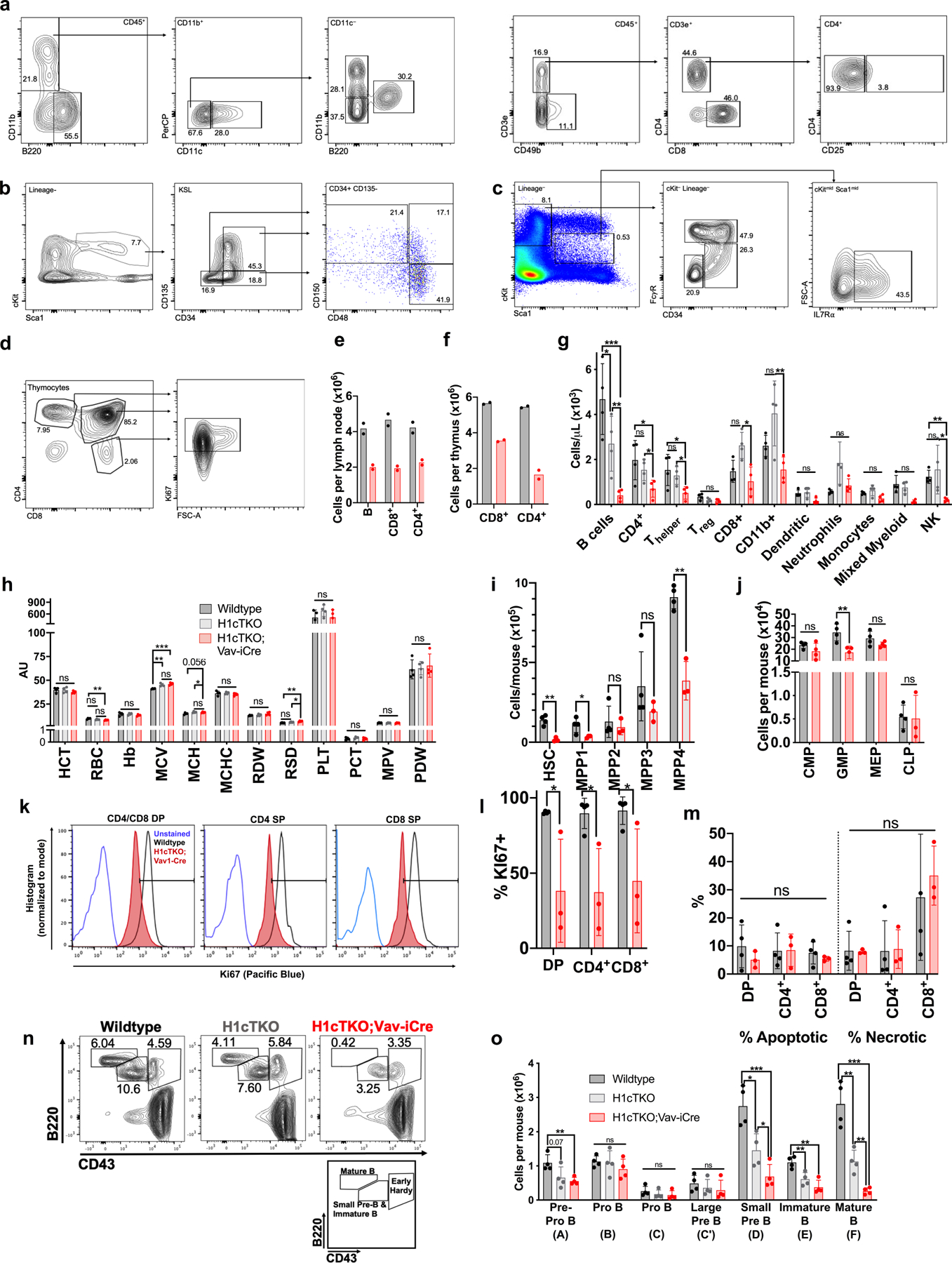
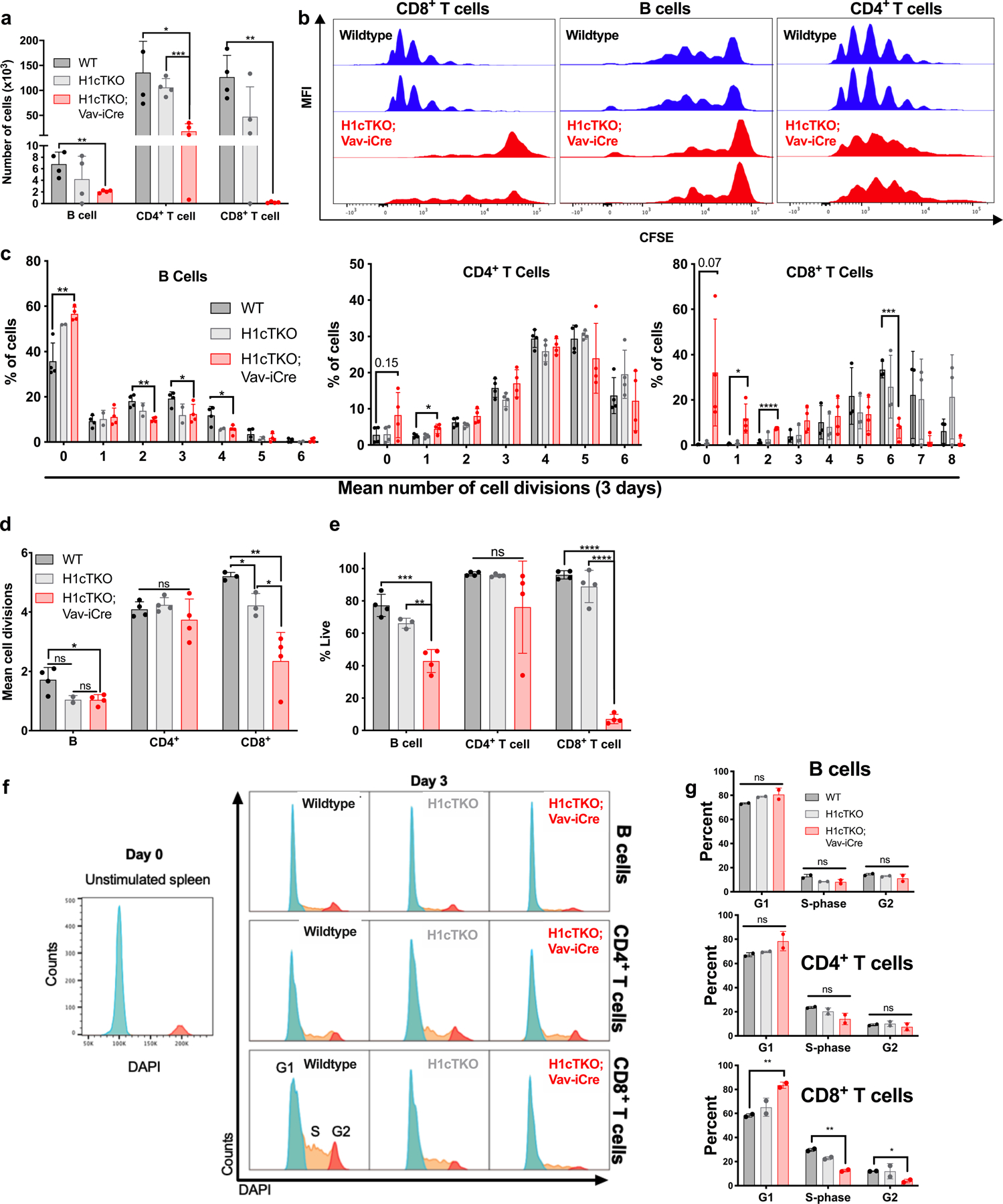
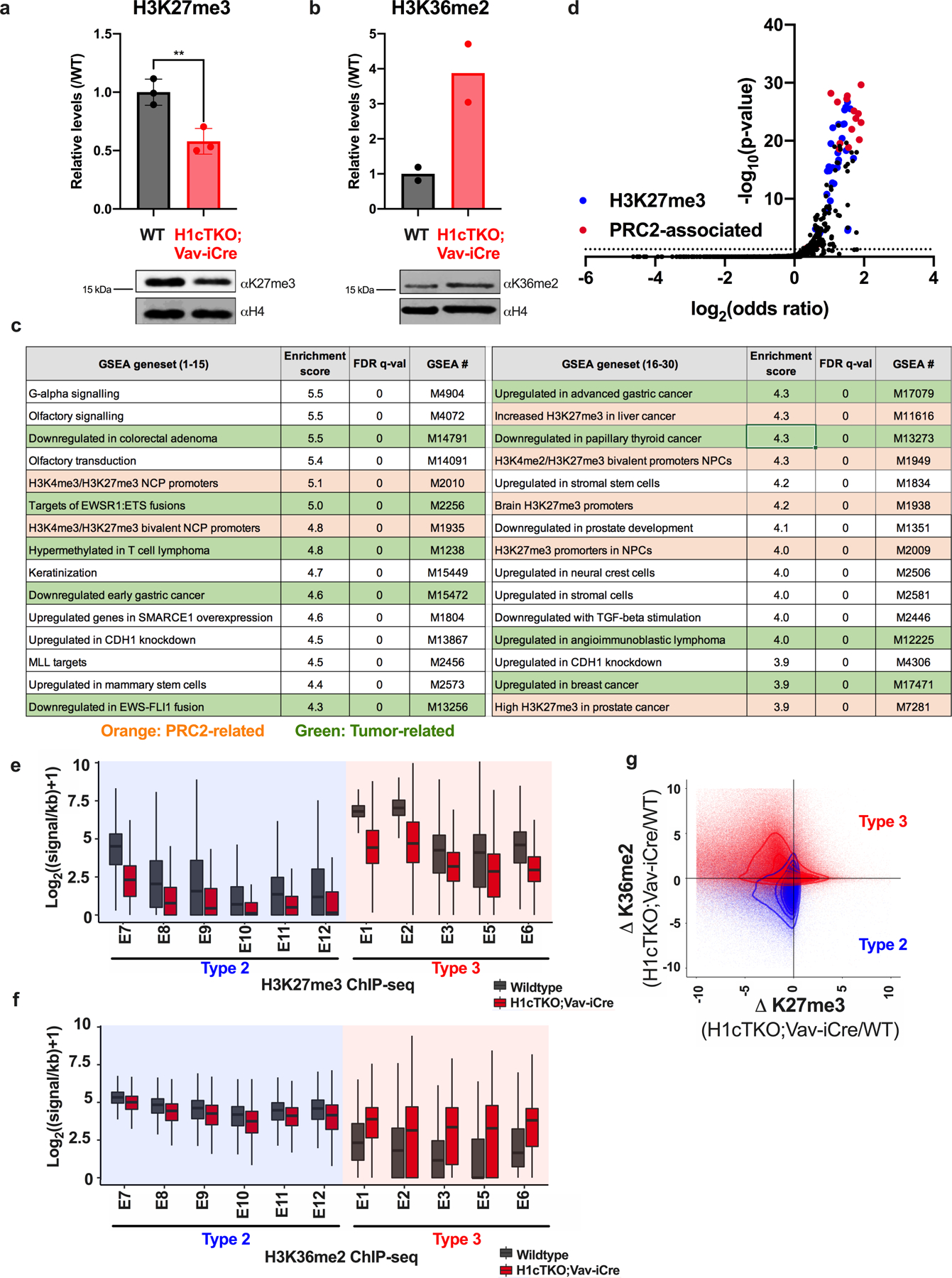
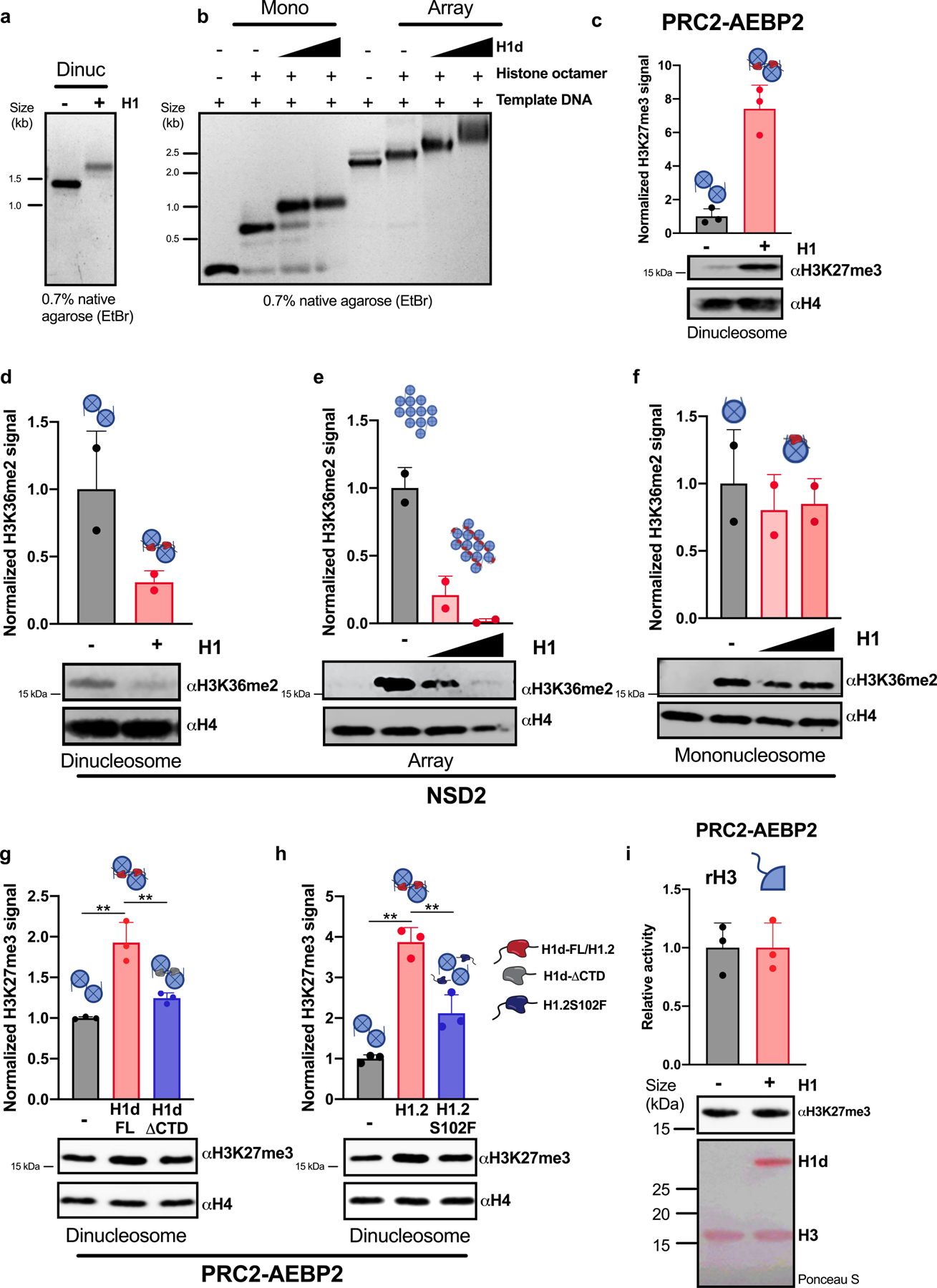
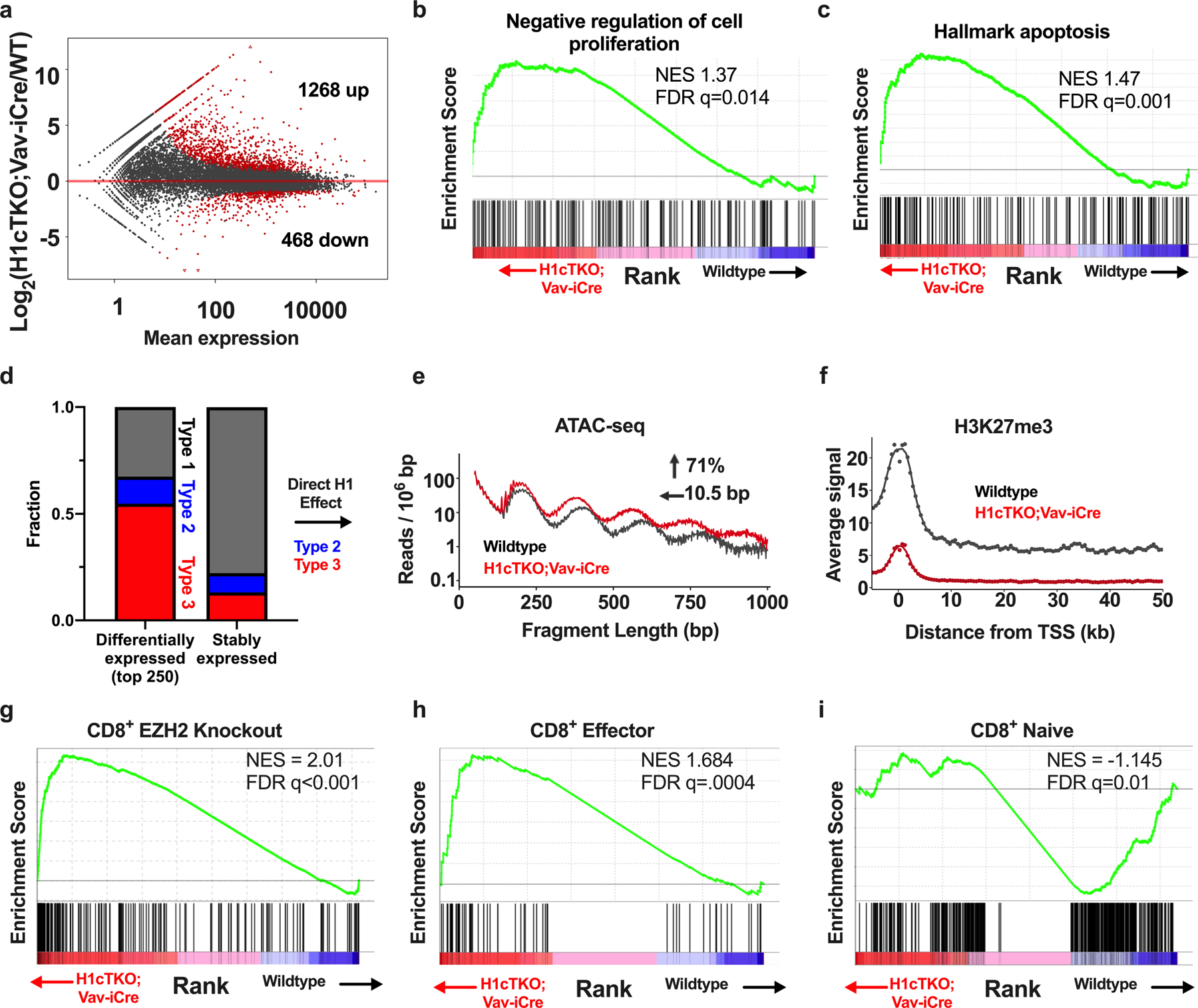
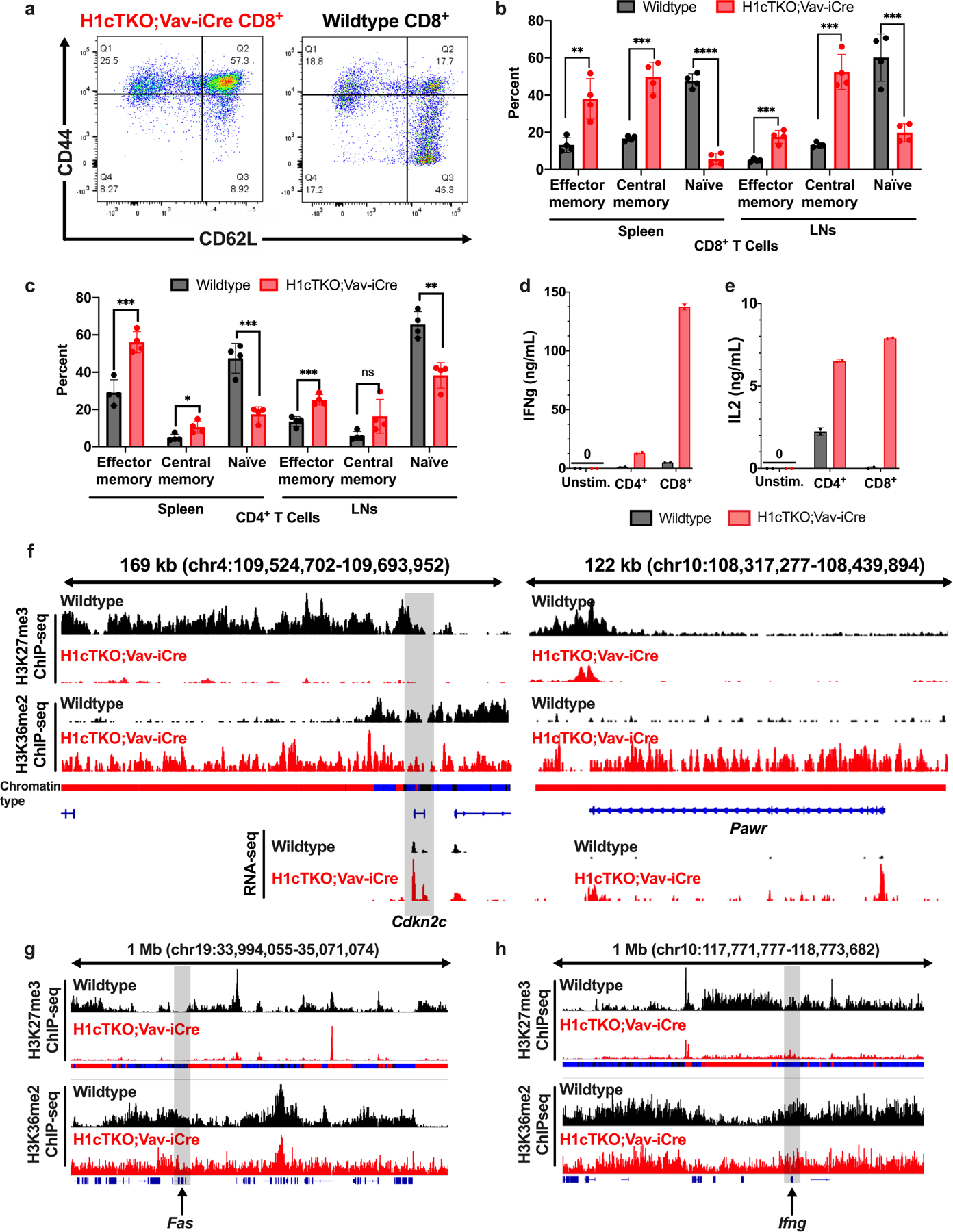
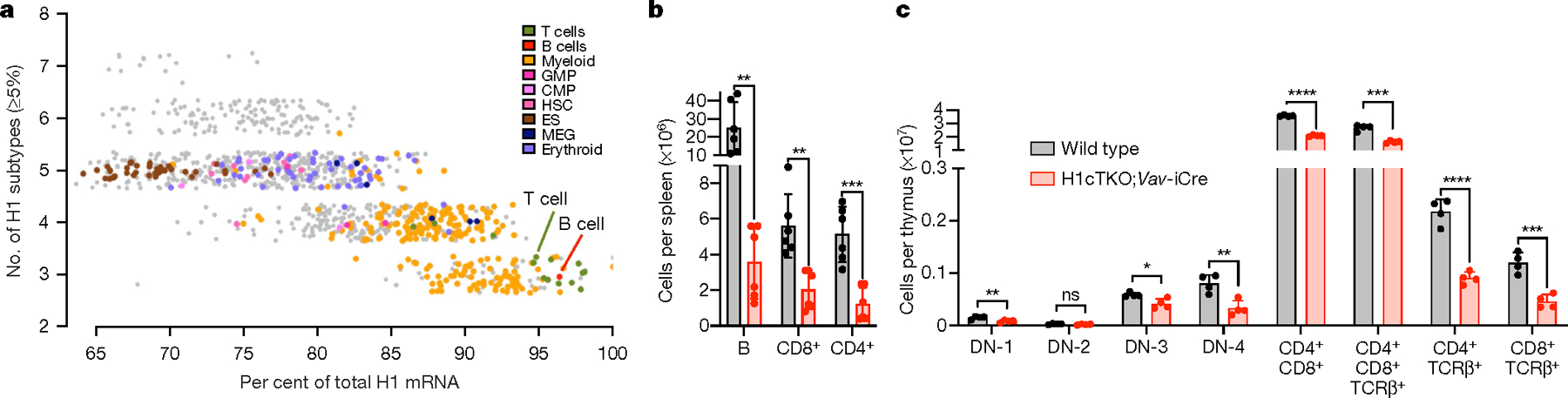
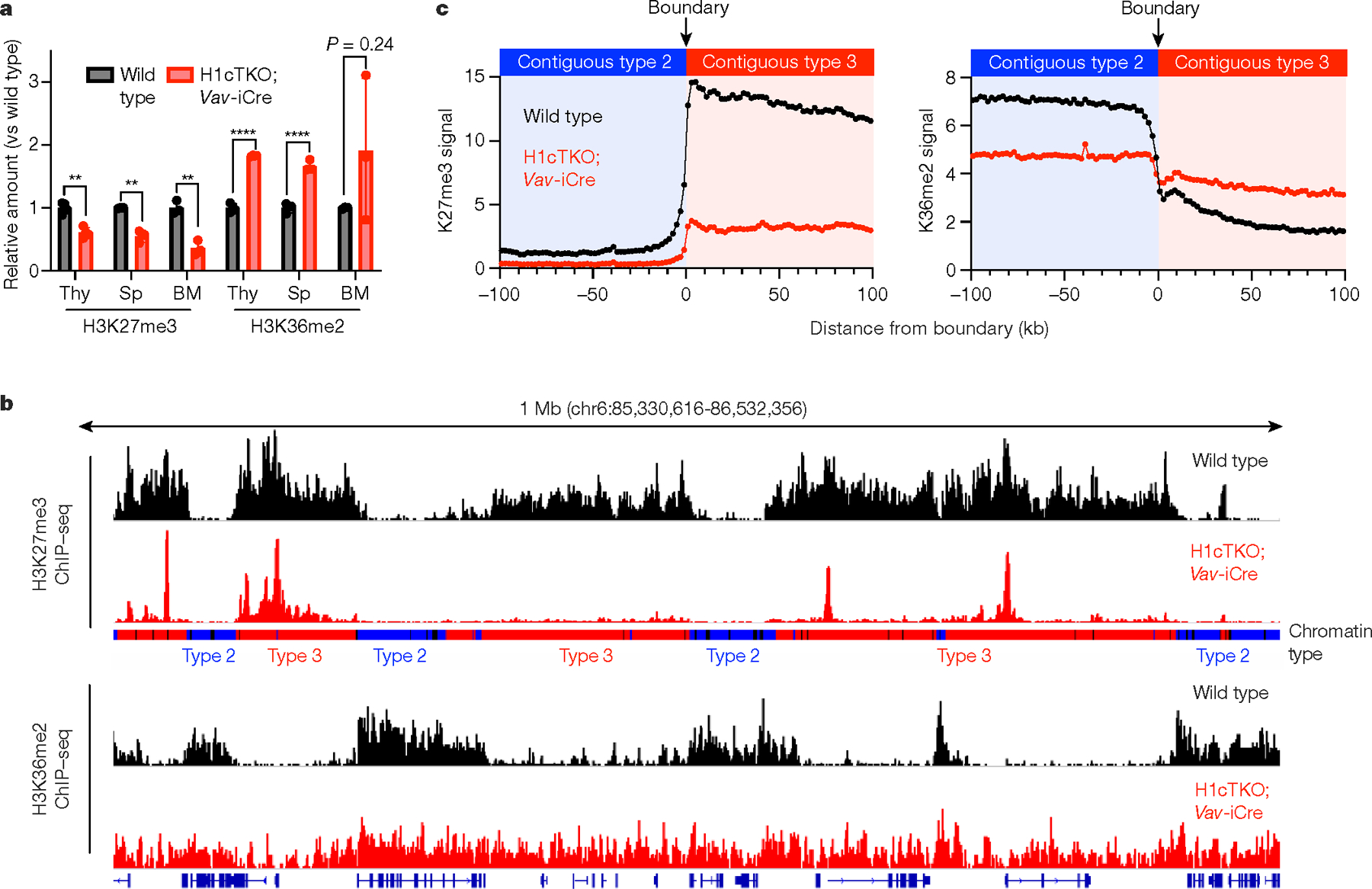
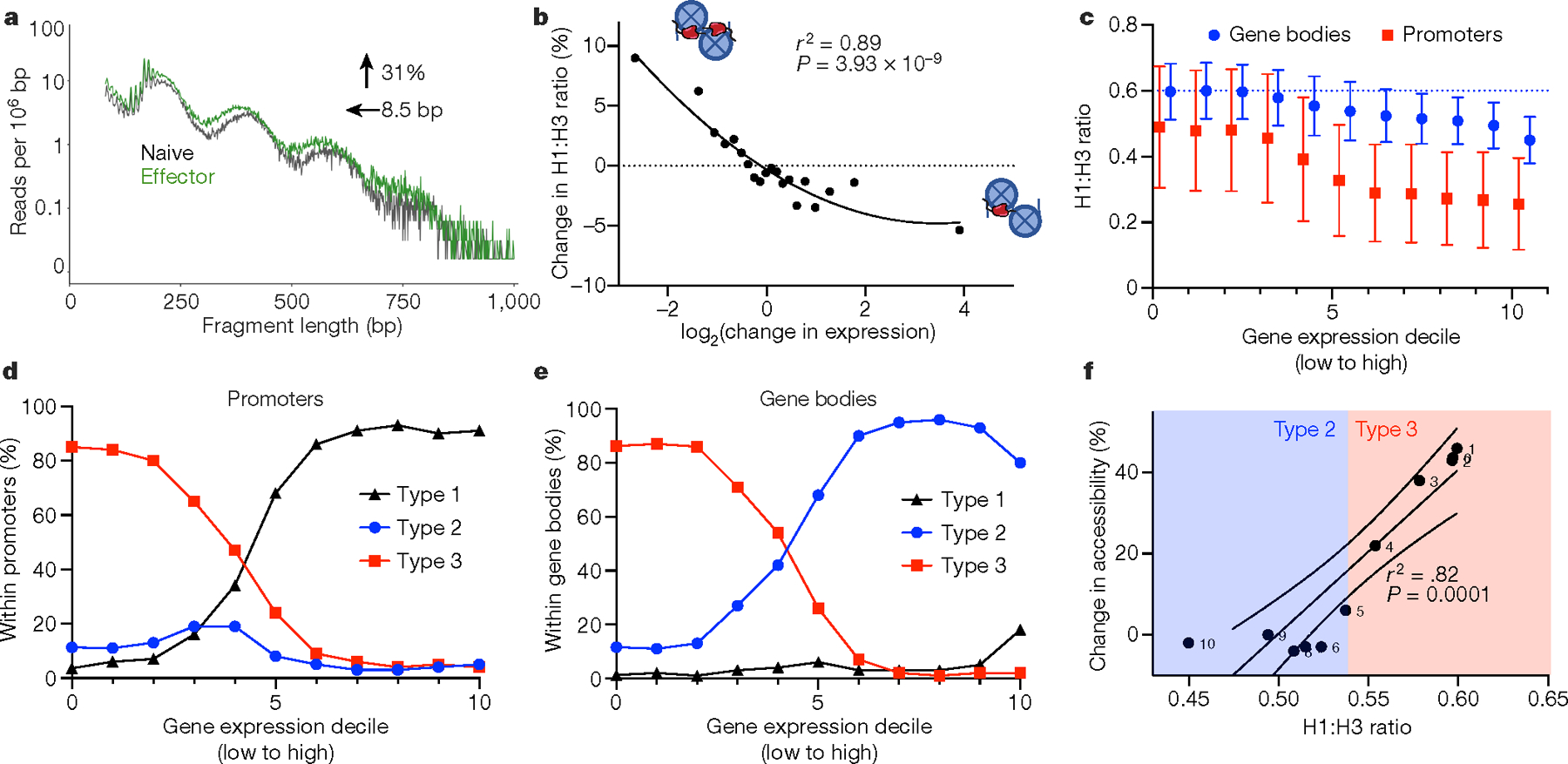
H1 linker histones are the most abundant chromatin-binding proteins1. In vitro studies indicate that their association with chromatin determines nucleosome spacing and enables arrays of nucleosomes to fold into more compact chromatin structures. However, the in vivo roles of H1 are poorly understood2. Here we show that the local density of H1 controls the balance of repressive and active chromatin domains by promoting genomic compaction. We generated a conditional triple-H1-knockout mouse strain and depleted H1 in haematopoietic cells. H1 depletion in T cells leads to de-repression of T cell activation genes, a process that mimics normal T cell activation. Comparison of chromatin structure in normal and H1-depleted CD8+ T cells reveals that H1-mediated chromatin compaction occurs primarily in regions of the genome containing higher than average levels of H1: the chromosome conformation capture (Hi-C) B compartment and regions of the Hi-C A compartment marked by PRC2. Reduction of H1 stoichiometry leads to decreased H3K27 methylation, increased H3K36 methylation, B-to-A-compartment shifting and an increase in interaction frequency between compartments. In vitro, H1 promotes PRC2-mediated H3K27 methylation and inhibits NSD2-mediated H3K36 methylation. Mechanistically, H1 mediates these opposite effects by promoting physical compaction of the chromatin substrate. Our results establish H1 as a critical regulator of gene silencing through localized control of chromatin compaction, 3D genome organization and the epigenetic landscape.
Conflict of interest statement
Figures





Comment in
-
Chromatin control by H1 histones.Nat Rev Genet. 2021 Feb;22(2):68-69. doi: 10.1038/s41576-020-00323-7. Nat Rev Genet. 2021. PMID: 33323999 No abstract available.
References
-
- Woodcock CL, Skoultchi AI & Fan Y Role of linker histone in chromatin structure and function: H1 stoichiometry and nucleosome repeat length. Chromosome Res 14, 17–25 (2006). - PubMed
-
- Fan Y et al. Histone H1 depletion in mammals alters global chromatin structure but causes specific changes in gene regulation. Cell 123, 1199–1212 (2005). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- GM110104/NH/NIH HHS/United States
- AI118891/NH/NIH HHS/United States
- P01 CA196539/CA/NCI NIH HHS/United States
- R01 CA234561/CA/NCI NIH HHS/United States
- P30 CA008748/CA/NCI NIH HHS/United States
- R01 AI118891/AI/NIAID NIH HHS/United States
- F30 DK107182/DK/NIDDK NIH HHS/United States
- T32 GM007288/GM/NIGMS NIH HHS/United States
- R01 CA226861/CA/NCI NIH HHS/United States
- P30 CA013330/CA/NCI NIH HHS/United States
- R01 GM116143/GM/NIGMS NIH HHS/United States
- F30 CA210539/CA/NCI NIH HHS/United States
- R35 GM138386/GM/NIGMS NIH HHS/United States
- F30 DK108532/DK/NIDDK NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
