

The functional proteome landscape of Escherichia coli
- PMID: 33299184
- PMCID: PMC7612278
- DOI: 10.1038/s41586-020-3002-5
The functional proteome landscape of Escherichia coli
Abstract
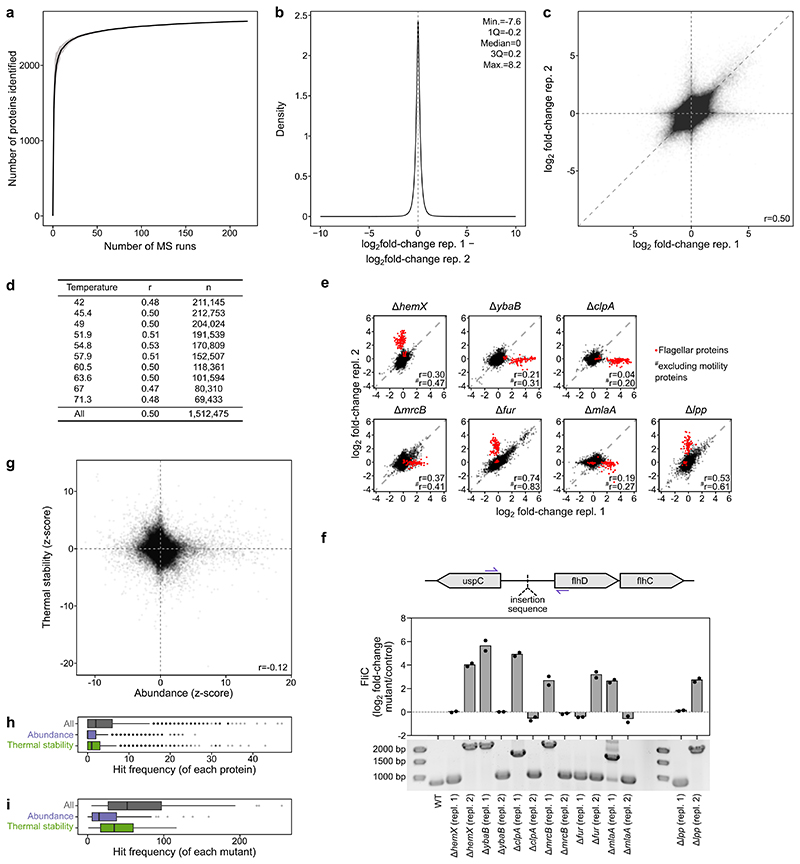
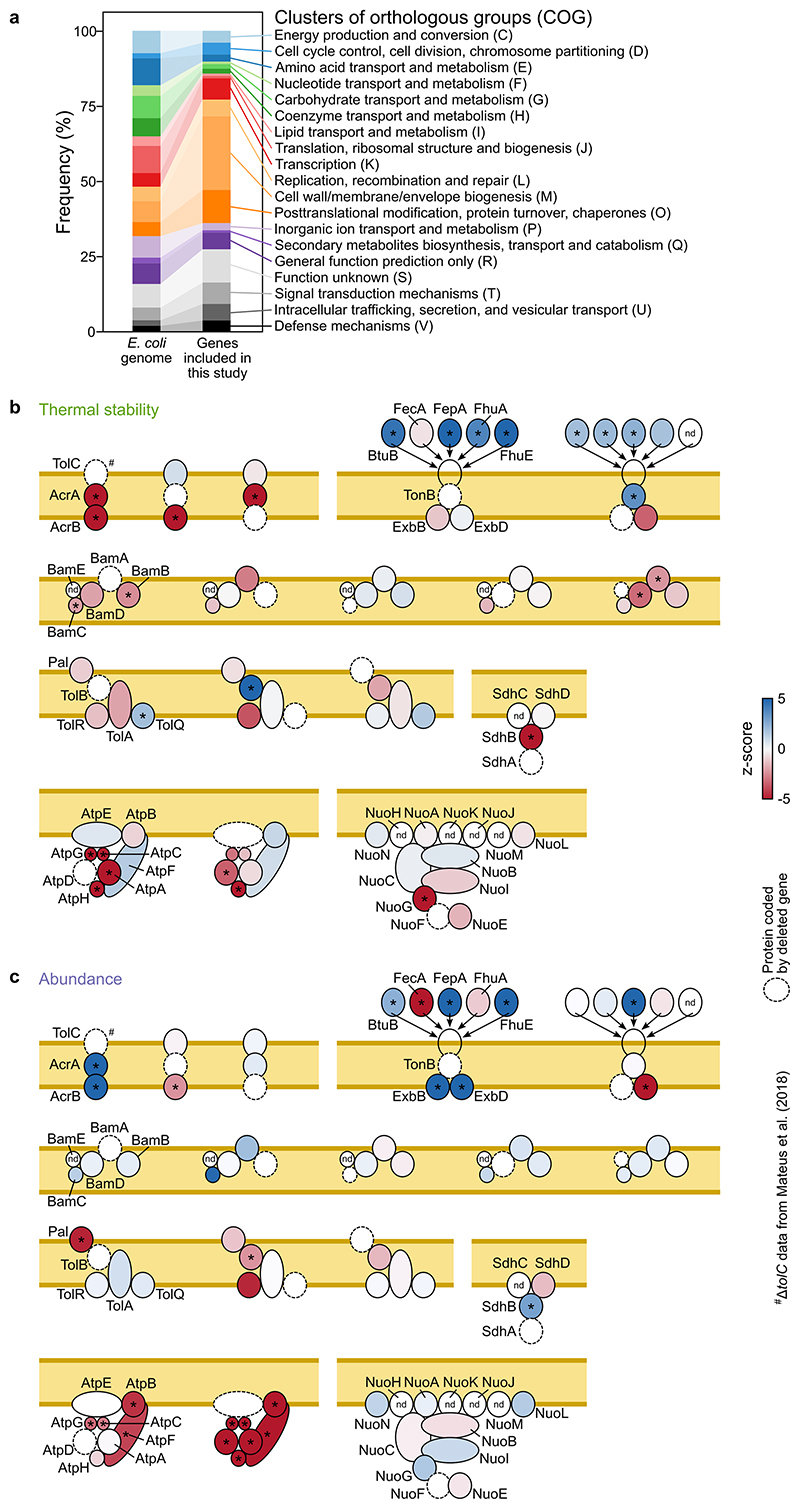
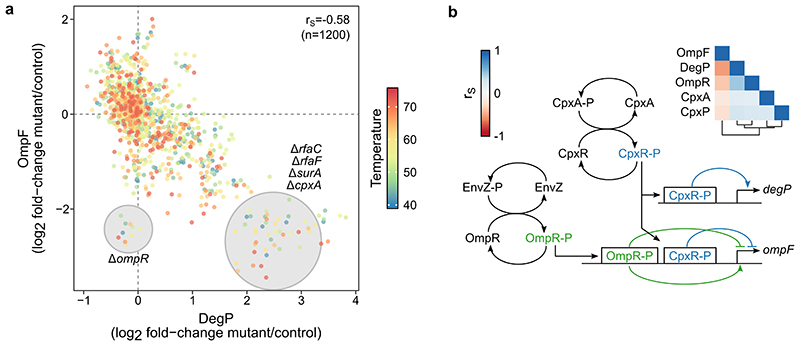
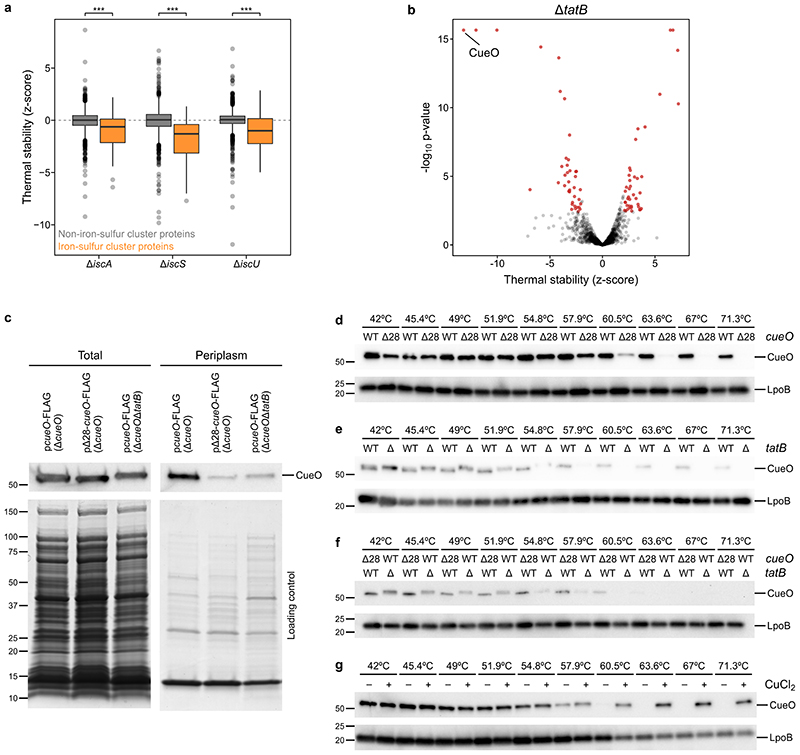
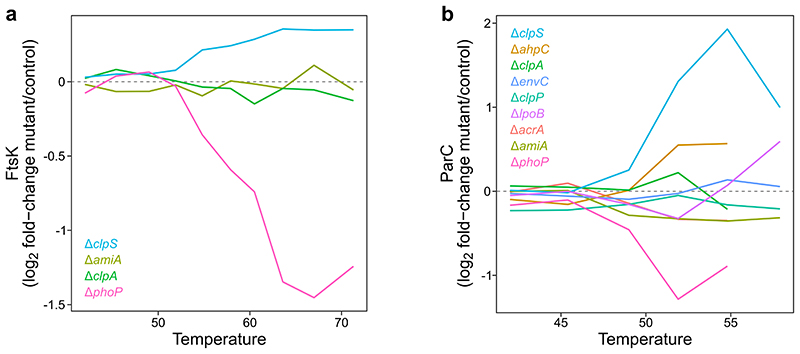
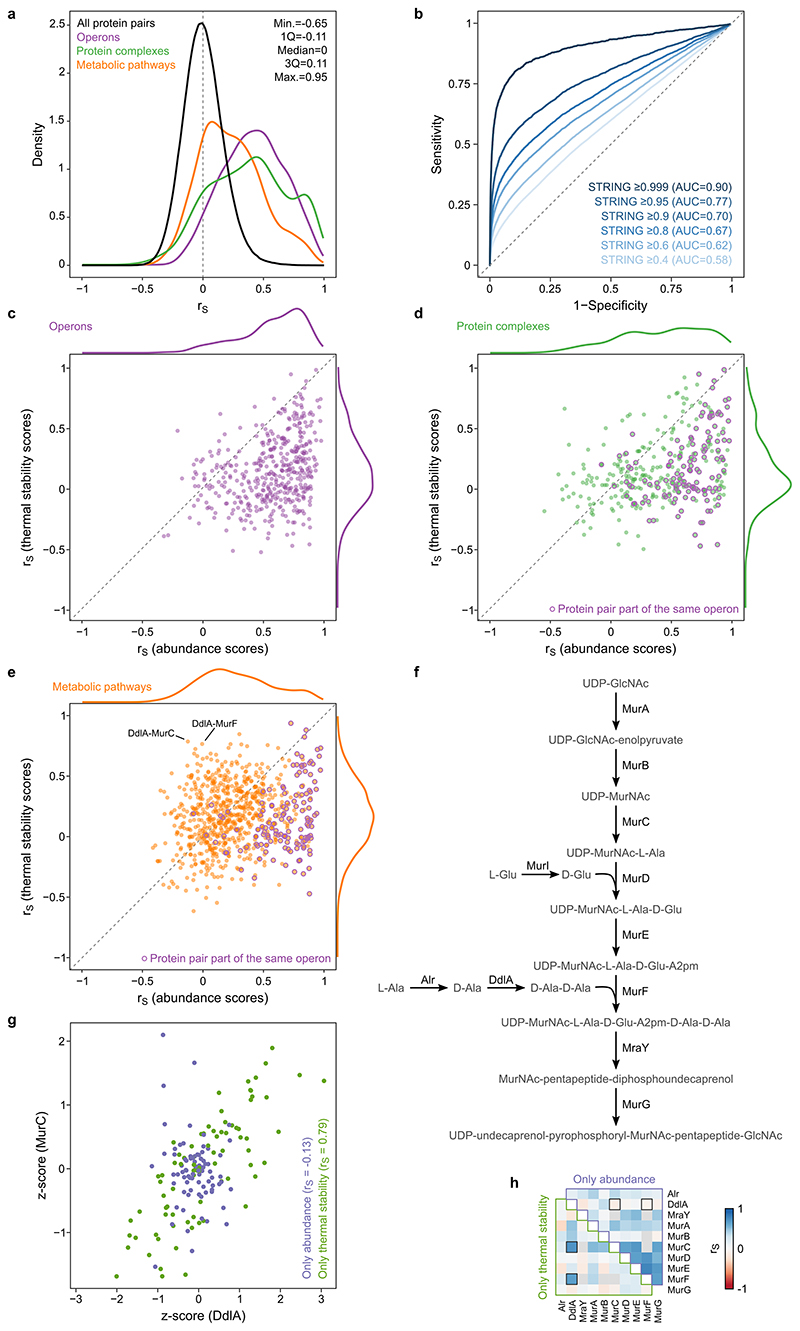
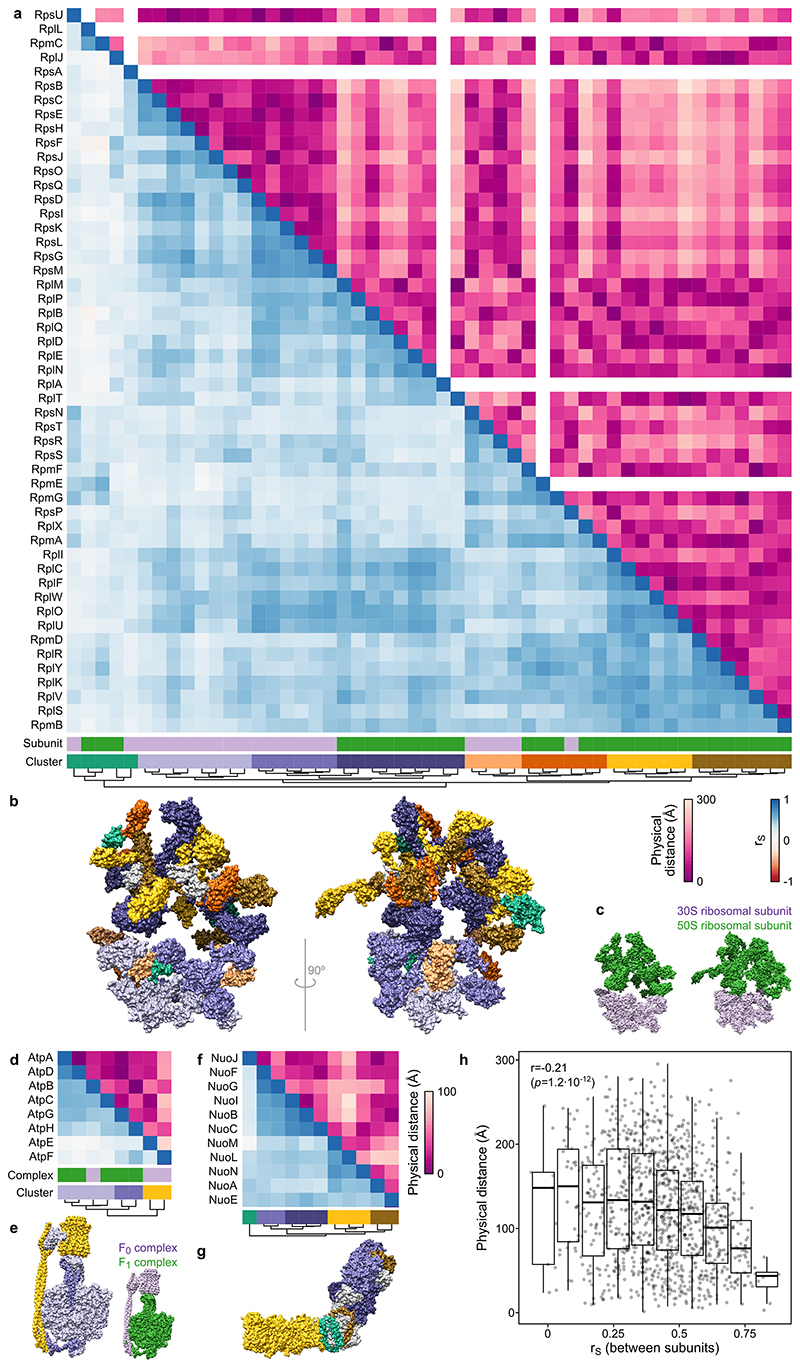
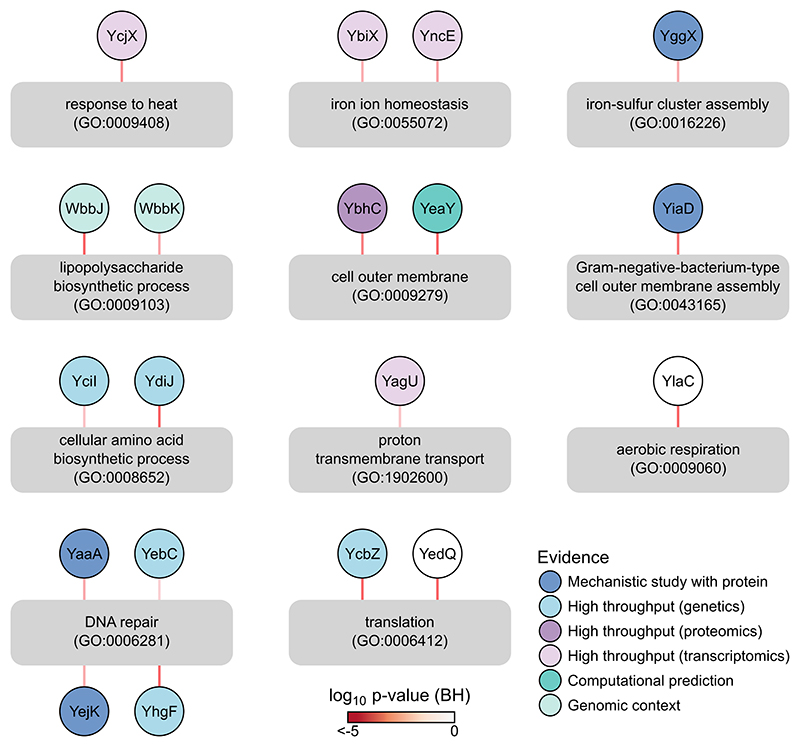
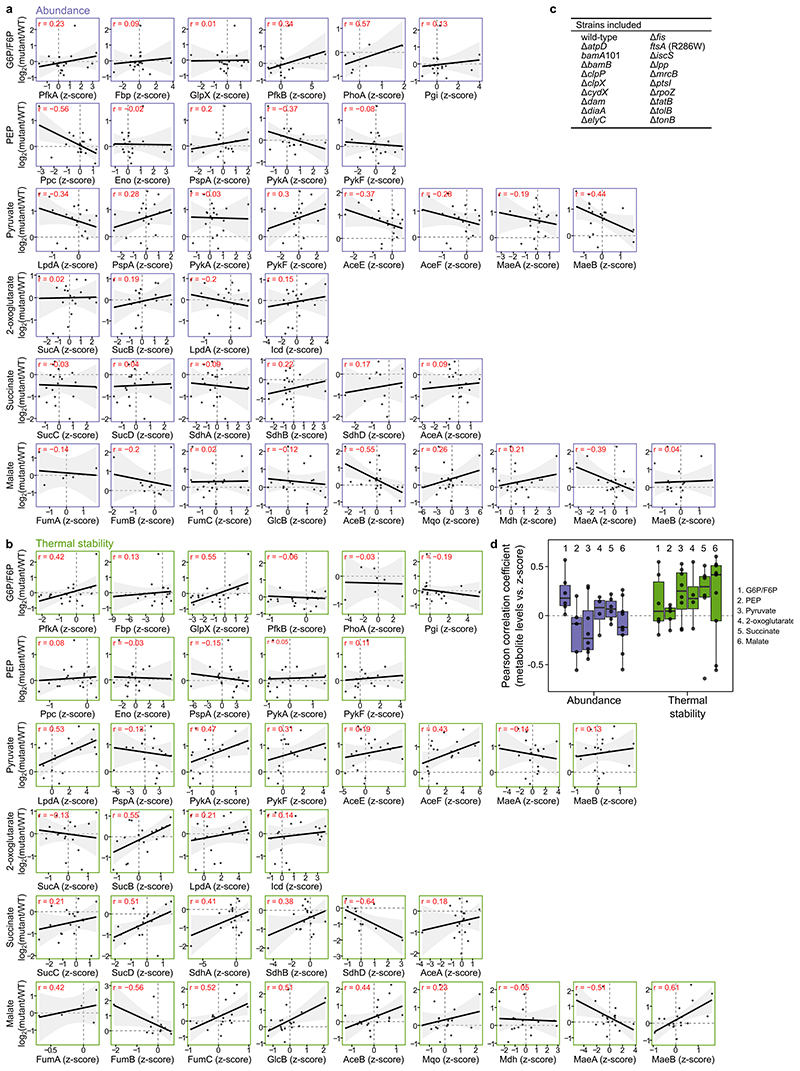
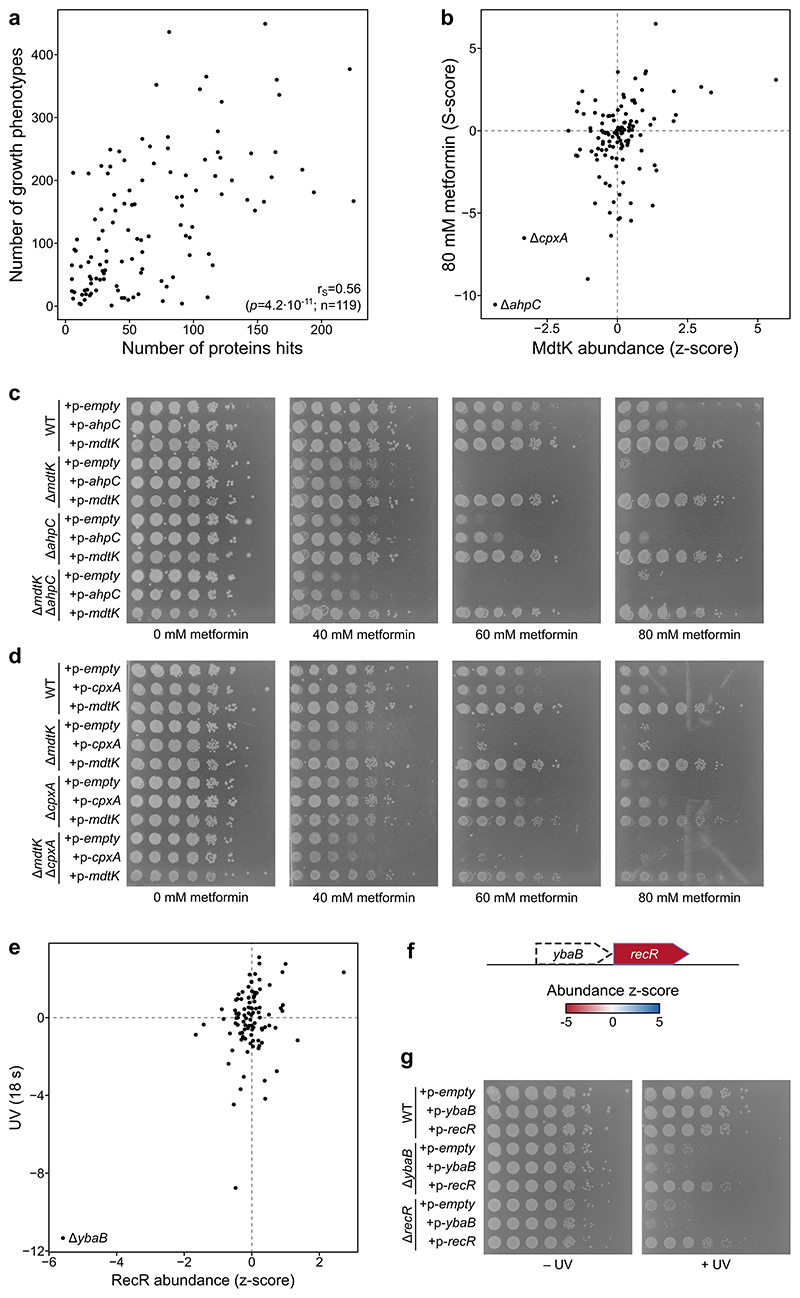
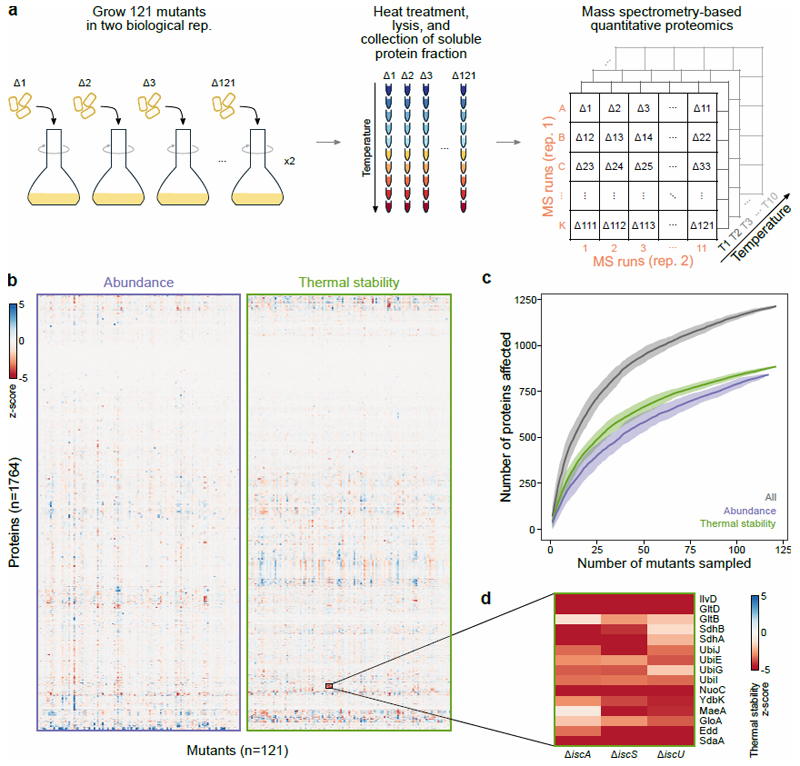
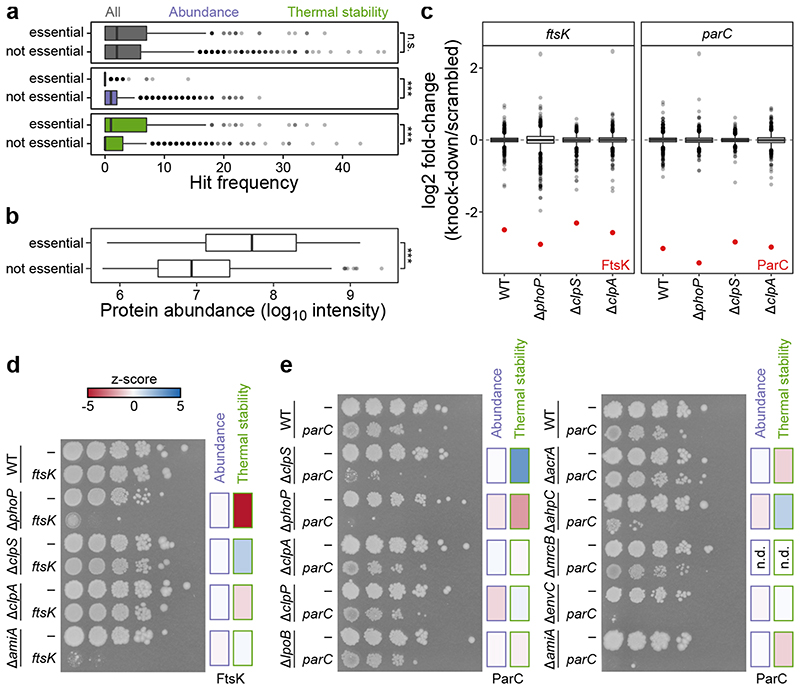
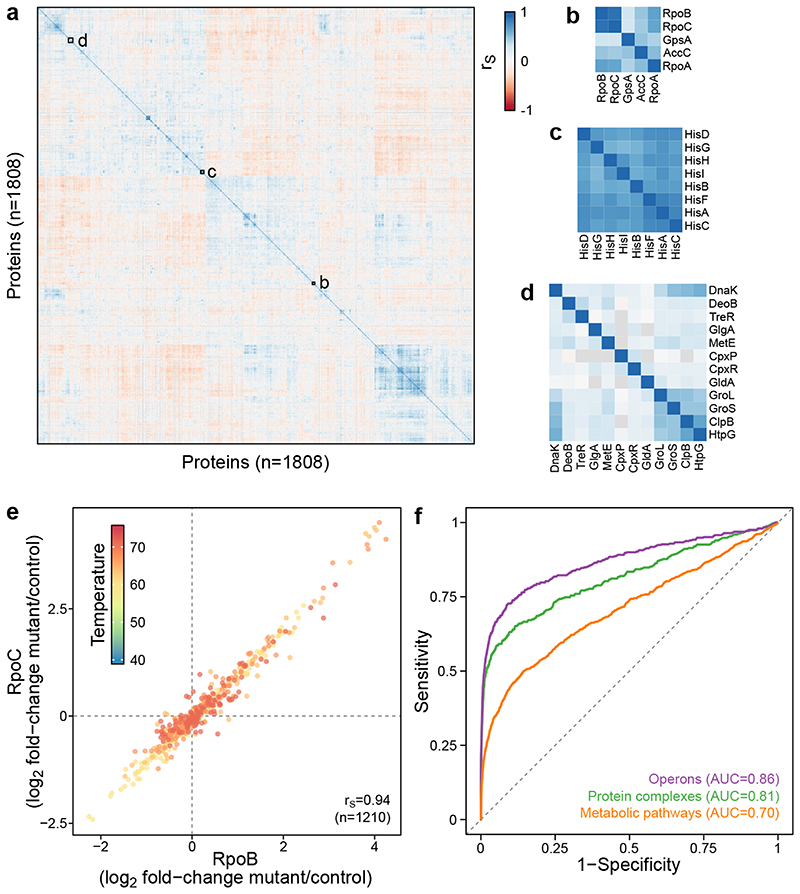
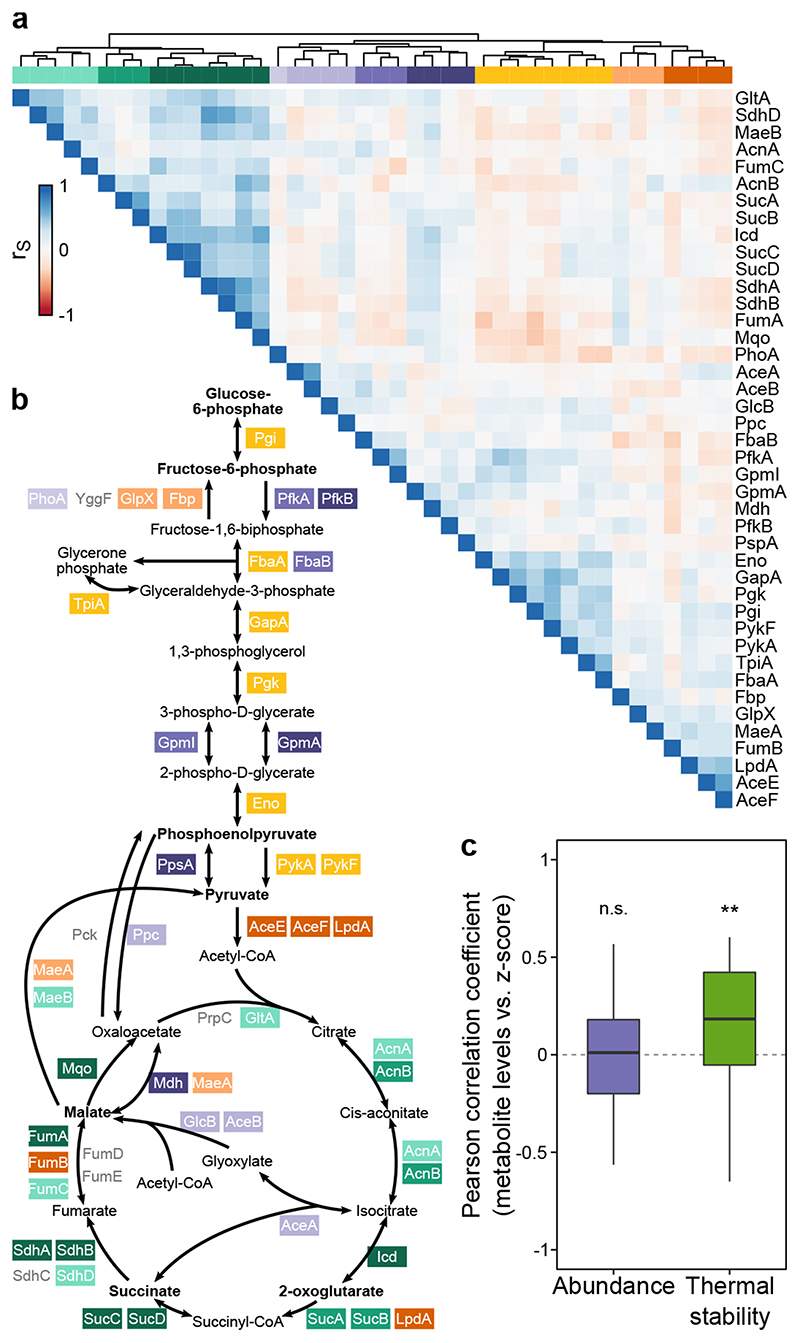
Recent developments in high-throughput reverse genetics1,2 have revolutionized our ability to map gene function and interactions3-6. The power of these approaches depends on their ability to identify functionally associated genes, which elicit similar phenotypic changes across several perturbations (chemical, environmental or genetic) when knocked out7-9. However, owing to the large number of perturbations, these approaches have been limited to growth or morphological readouts10. Here we use a high-content biochemical readout, thermal proteome profiling11, to measure the proteome-wide protein abundance and thermal stability in response to 121 genetic perturbations in Escherichia coli. We show that thermal stability, and therefore the state and interactions of essential proteins, is commonly modulated, raising the possibility of studying a protein group that is particularly inaccessible to genetics. We find that functionally associated proteins have coordinated changes in abundance and thermal stability across perturbations, owing to their co-regulation and physical interactions (with proteins, metabolites or cofactors). Finally, we provide mechanistic insights into previously determined growth phenotypes12 that go beyond the deleted gene. These data represent a rich resource for inferring protein functions and interactions.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
