

A metastasis map of human cancer cell lines
- PMID: 33299191
- PMCID: PMC8439149
- DOI: 10.1038/s41586-020-2969-2
A metastasis map of human cancer cell lines
Erratum in
-
Publisher Correction: A metastasis map of human cancer cell lines.Nature. 2021 Nov;599(7885):E7. doi: 10.1038/s41586-021-04149-z. Nature. 2021. PMID: 34732899 Free PMC article. No abstract available.
Abstract
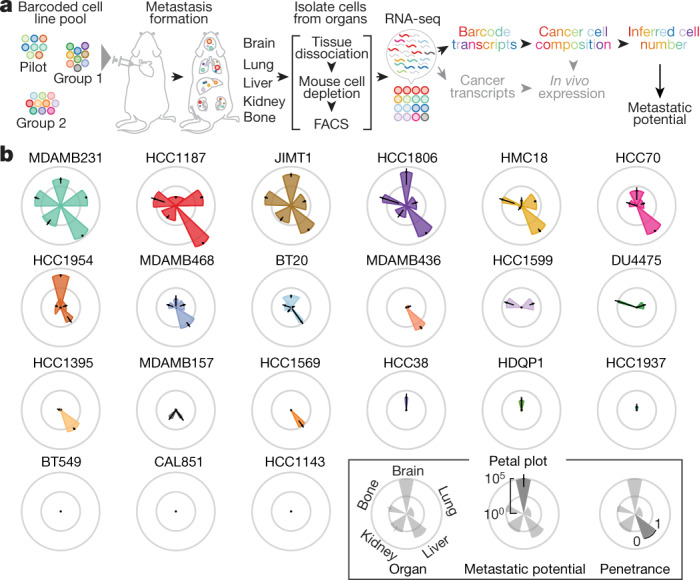
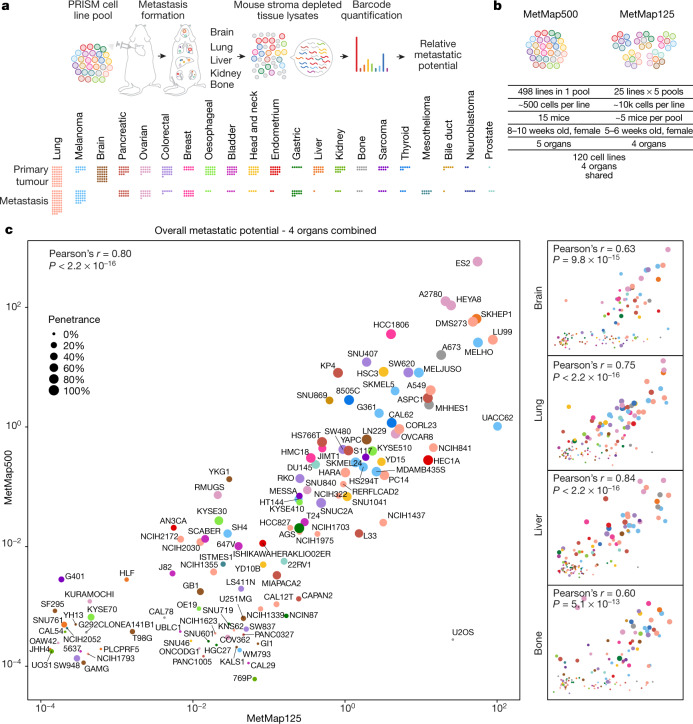
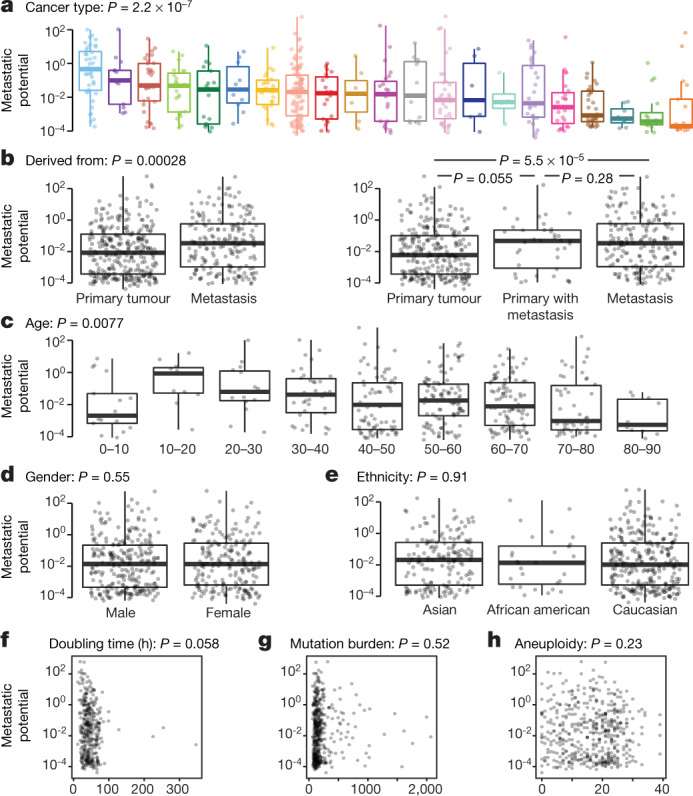
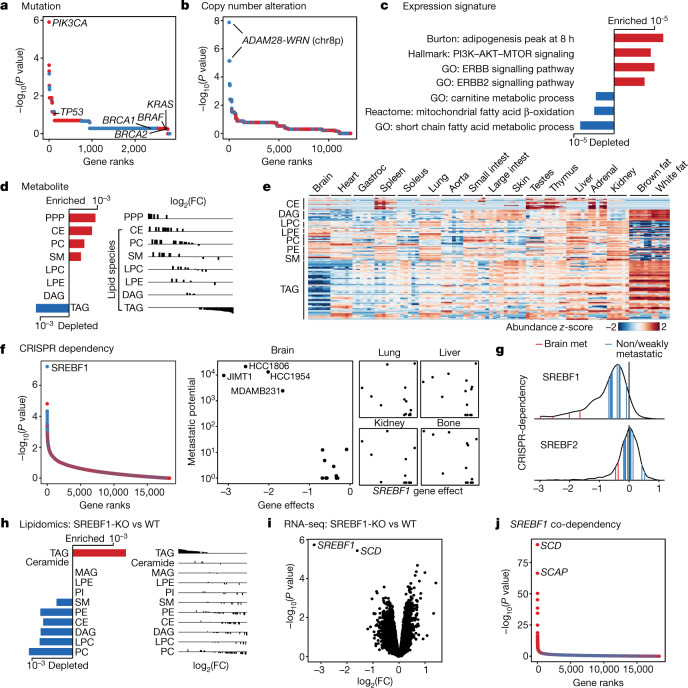
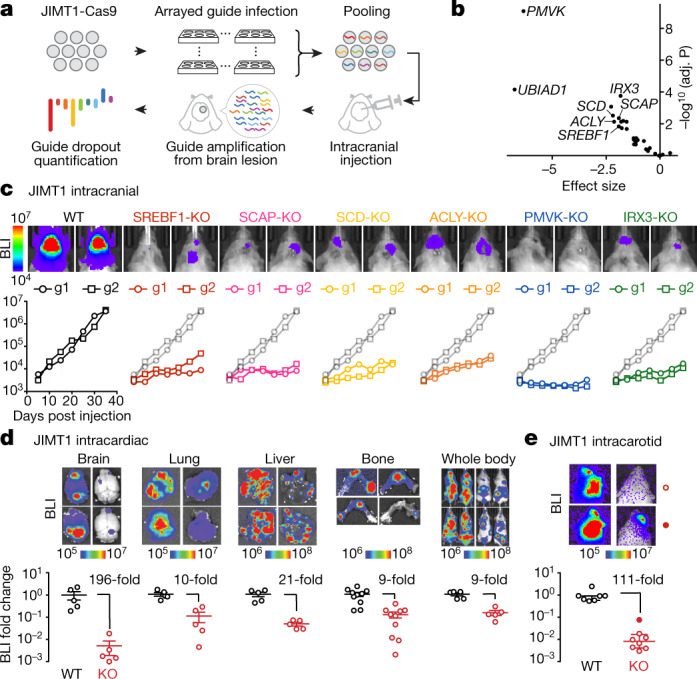
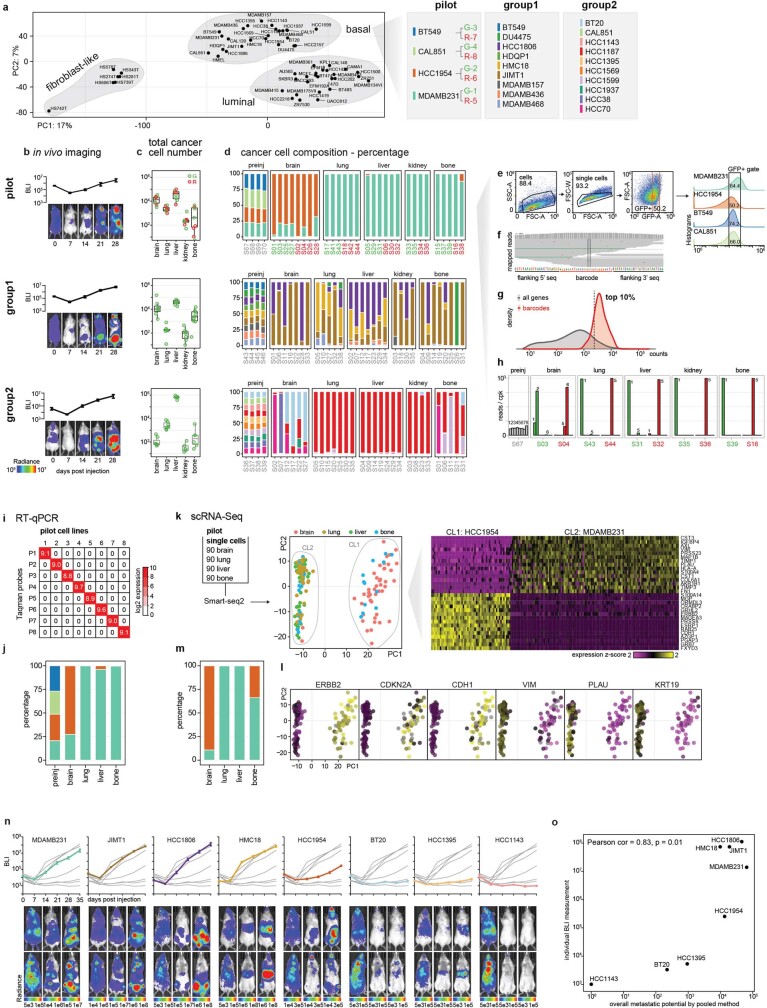
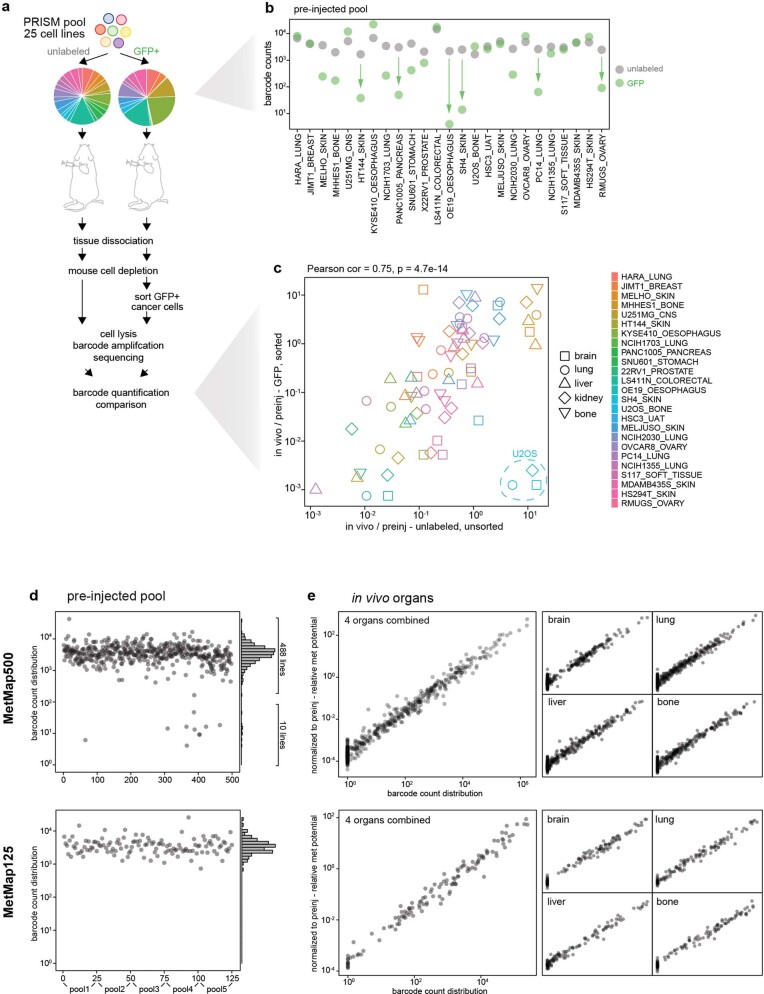
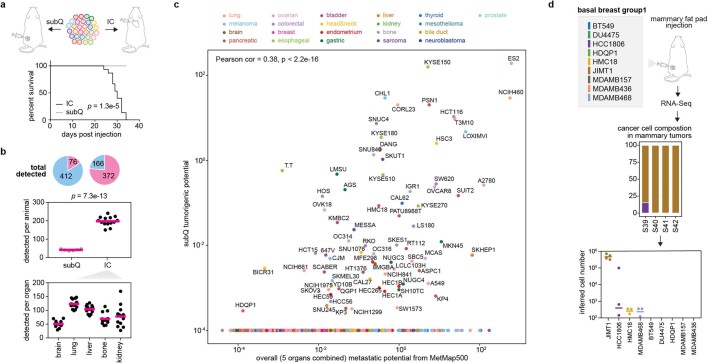
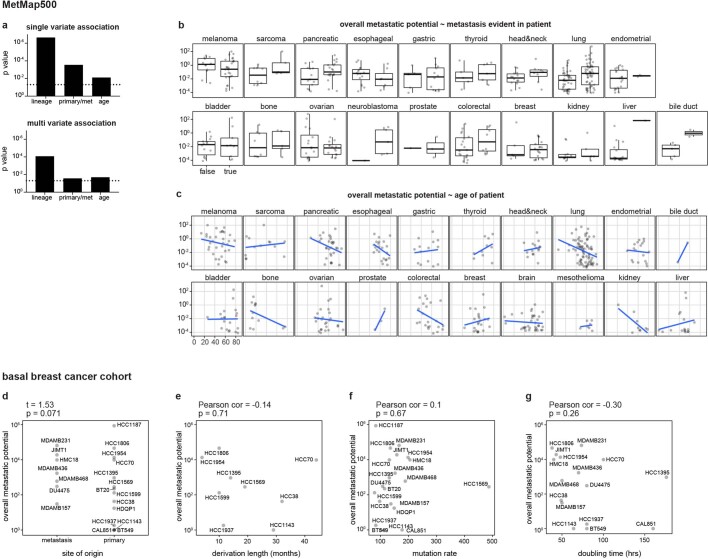
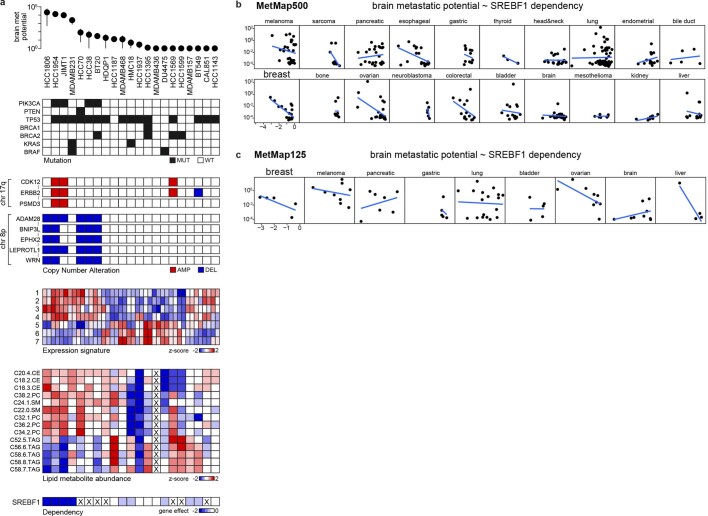
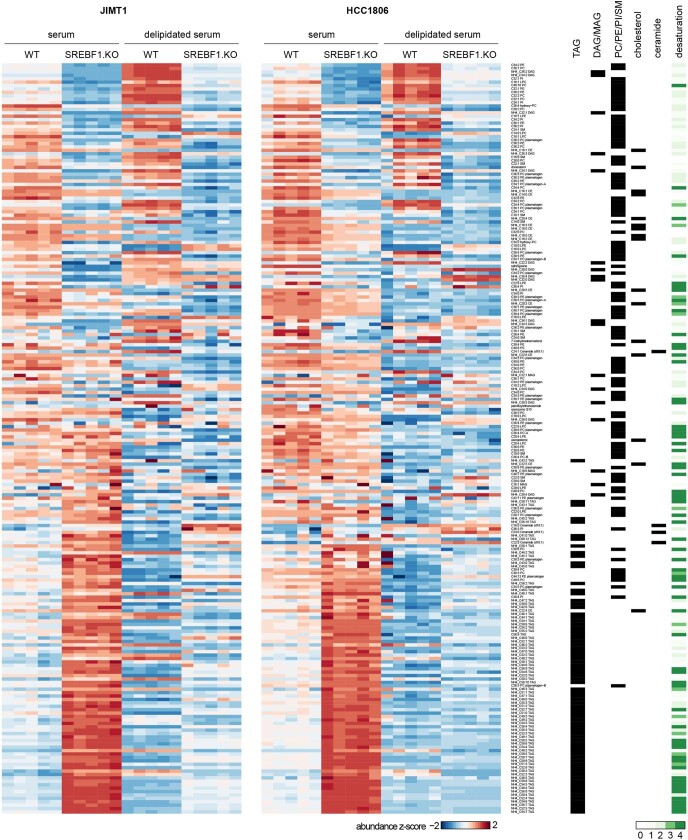
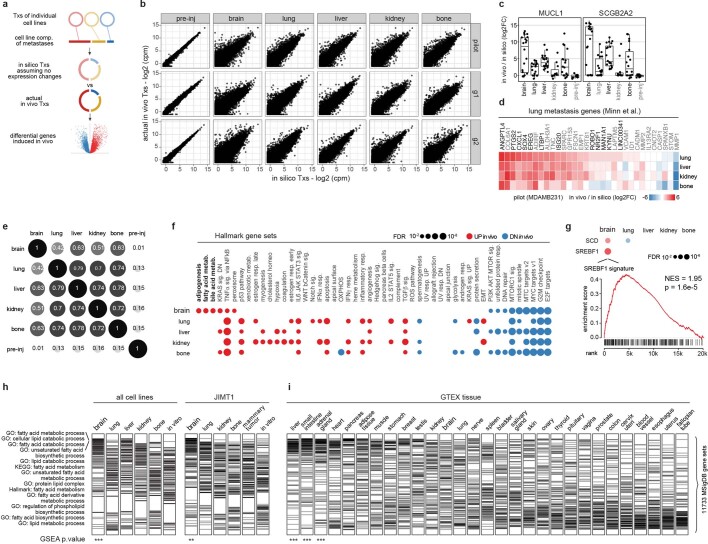
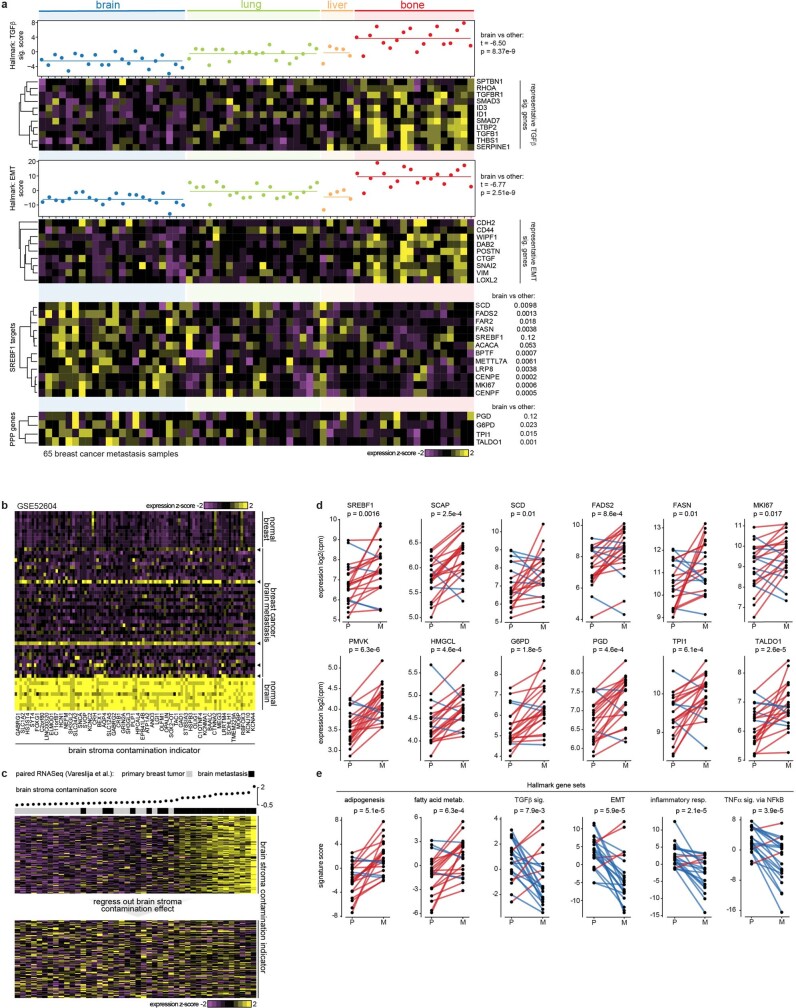
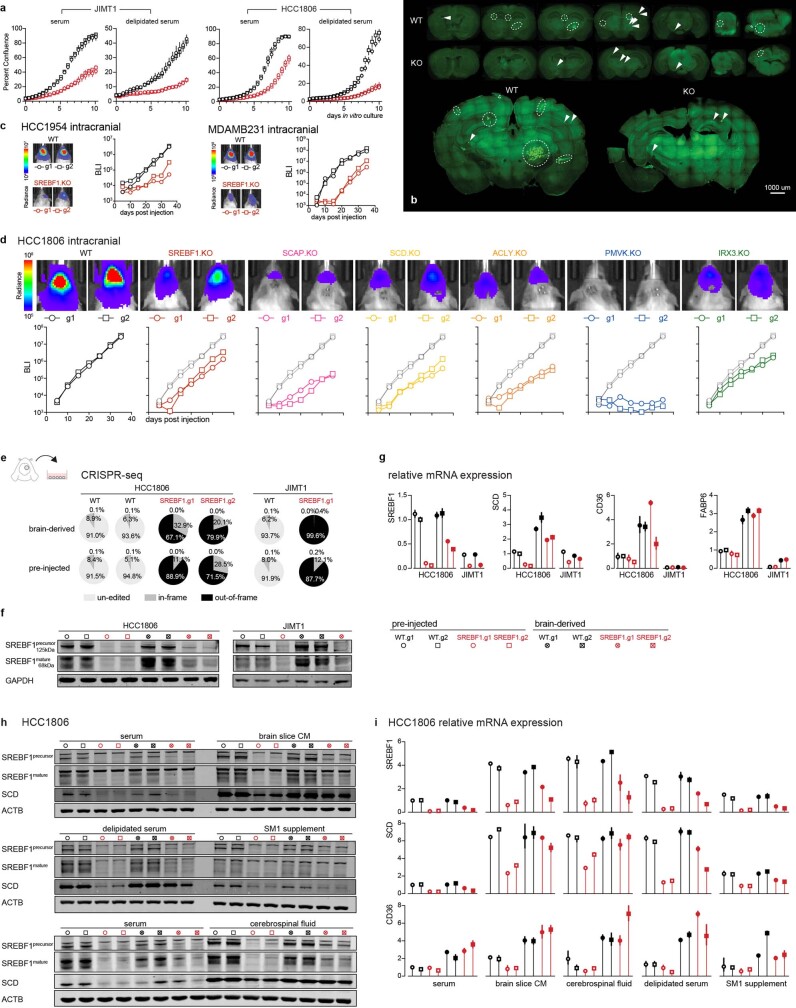
Most deaths from cancer are explained by metastasis, and yet large-scale metastasis research has been impractical owing to the complexity of in vivo models. Here we introduce an in vivo barcoding strategy that is capable of determining the metastatic potential of human cancer cell lines in mouse xenografts at scale. We validated the robustness, scalability and reproducibility of the method and applied it to 500 cell lines1,2 spanning 21 types of solid tumour. We created a first-generation metastasis map (MetMap) that reveals organ-specific patterns of metastasis, enabling these patterns to be associated with clinical and genomic features. We demonstrate the utility of MetMap by investigating the molecular basis of breast cancers capable of metastasizing to the brain-a principal cause of death in patients with this type of cancer. Breast cancers capable of metastasizing to the brain showed evidence of altered lipid metabolism. Perturbation of lipid metabolism in these cells curbed brain metastasis development, suggesting a therapeutic strategy to combat the disease and demonstrating the utility of MetMap as a resource to support metastasis research.
Conflict of interest statement
T.R.G. receives research funding unrelated to this project from Bayer HealthCare, Novo Ventures and Calico Life Sciences; holds equity in FORMA Therapeutics; is a consultant to GlaxoSmithKline; and is a founder of Sherlock Biosciences. M.G.V.H. is a scientific advisory board member for Agios Pharmaceuticals, Aeglea Biotherapeutics, Auron Therapeutics and iTeos Therapeutics. R.K.J. received a honorarium from Amgen; consultant fees from Chugai, Merck, Ophthotech, Pfizer, SPARC and SynDevRx; owns equity in Accurius, Enlight, Ophthotech and SynDevRx; and serves on the Boards of Trustees of Tekla Healthcare Investors, Tekla Life Sciences Investors, Tekla Healthcare Opportunities Fund and Tekla World Healthcare Fund. No reagents or funding from these organizations were used in this study. X.J. and T.R.G. are named as inventors on pending PCT Patent Application No. PCT/US20/29584 filed by The Broad Institute, which describes compositions and methods for characterizing the metastatic potential of cancer cell lines. The other authors declare no competing interests.
Figures

Comment in
-
Heterogeneity in Metastatic Potential of Cancer Cells Is Revealed En Masse.Cancer Cell. 2021 Feb 8;39(2):148-150. doi: 10.1016/j.ccell.2021.01.017. Cancer Cell. 2021. PMID: 33561396
-
Fatty acid synthesis enables brain metastasis.Nat Cancer. 2021 Apr;2(4):374-376. doi: 10.1038/s43018-021-00202-y. Nat Cancer. 2021. PMID: 35122002 No abstract available.
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
