

The integrated stress response mediates necrosis in murine Mycobacterium tuberculosis granulomas
- PMID: 33301427
- PMCID: PMC7843230
- DOI: 10.1172/JCI130319
The integrated stress response mediates necrosis in murine Mycobacterium tuberculosis granulomas
Abstract
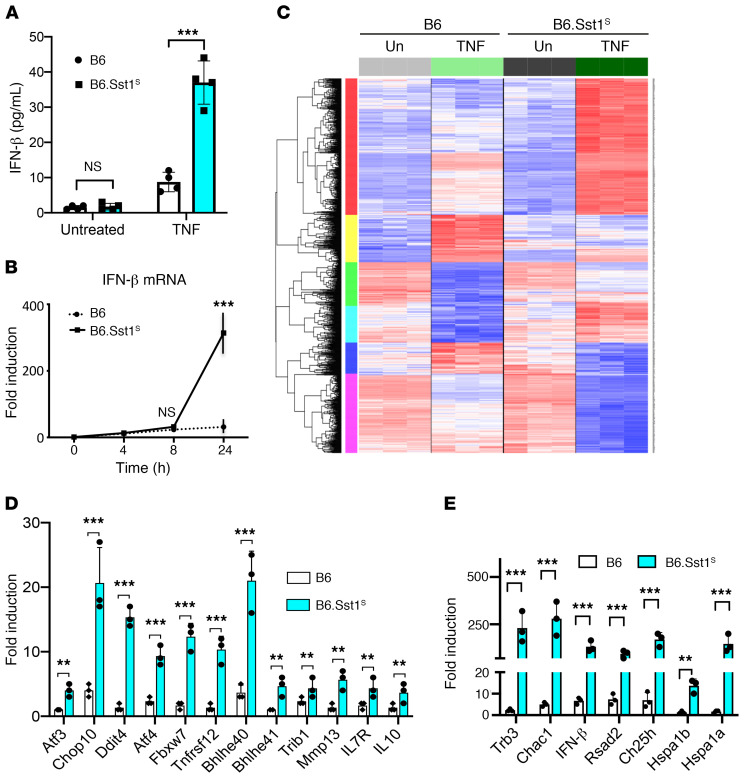
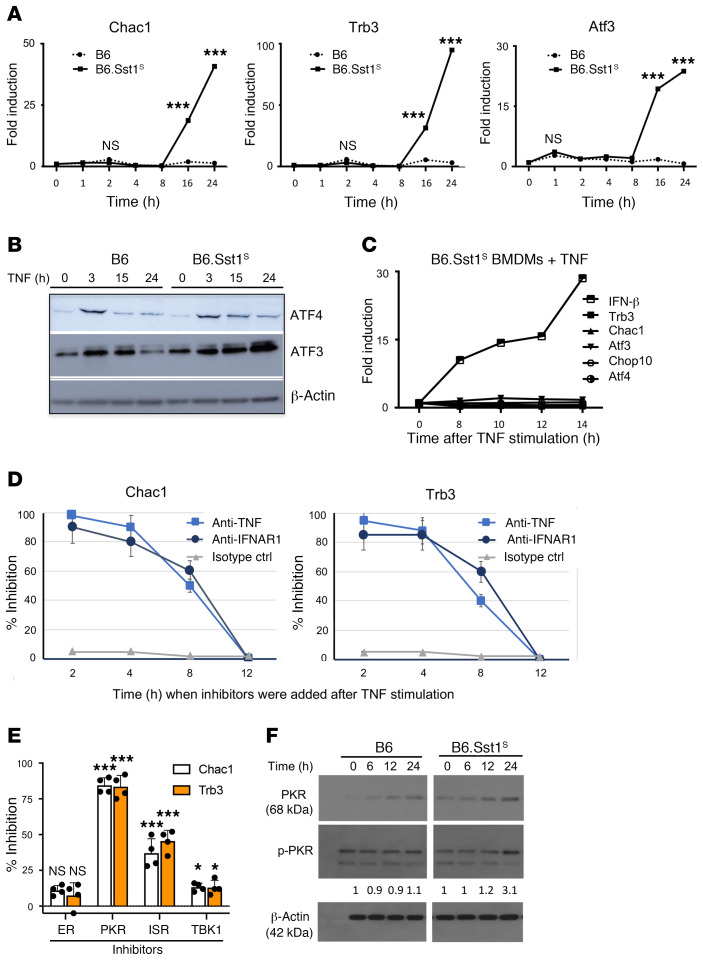
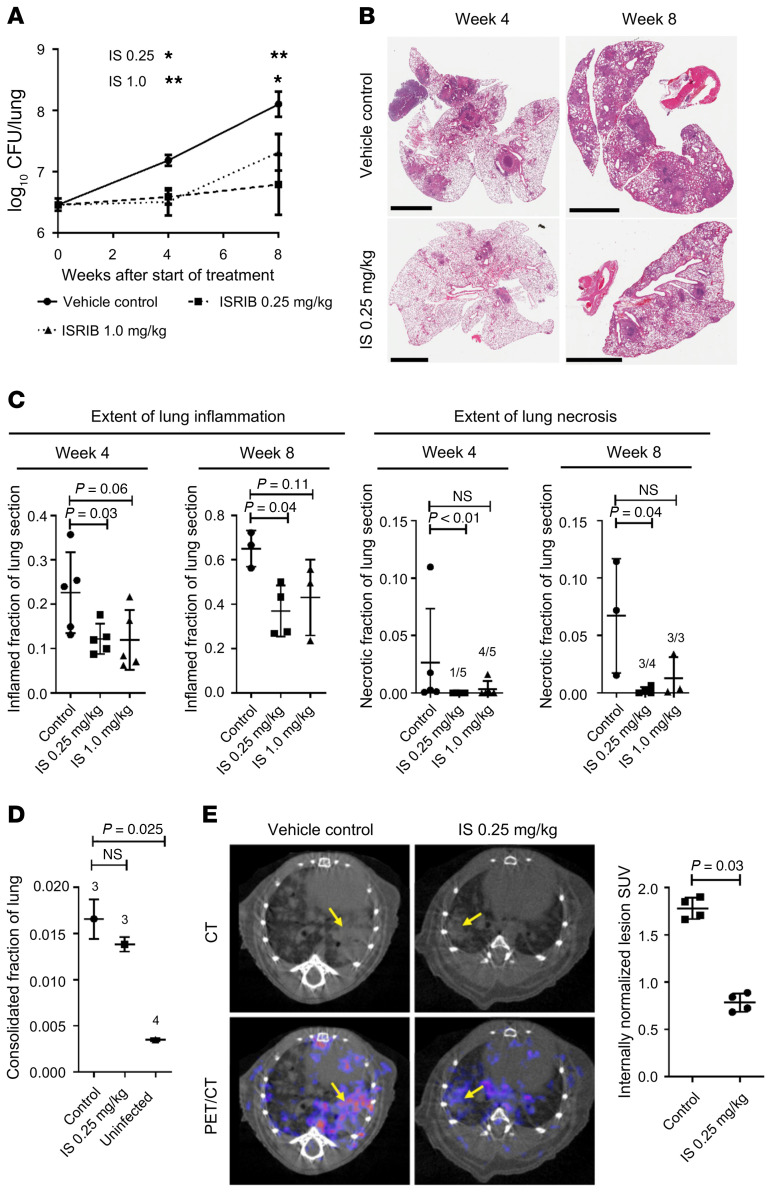
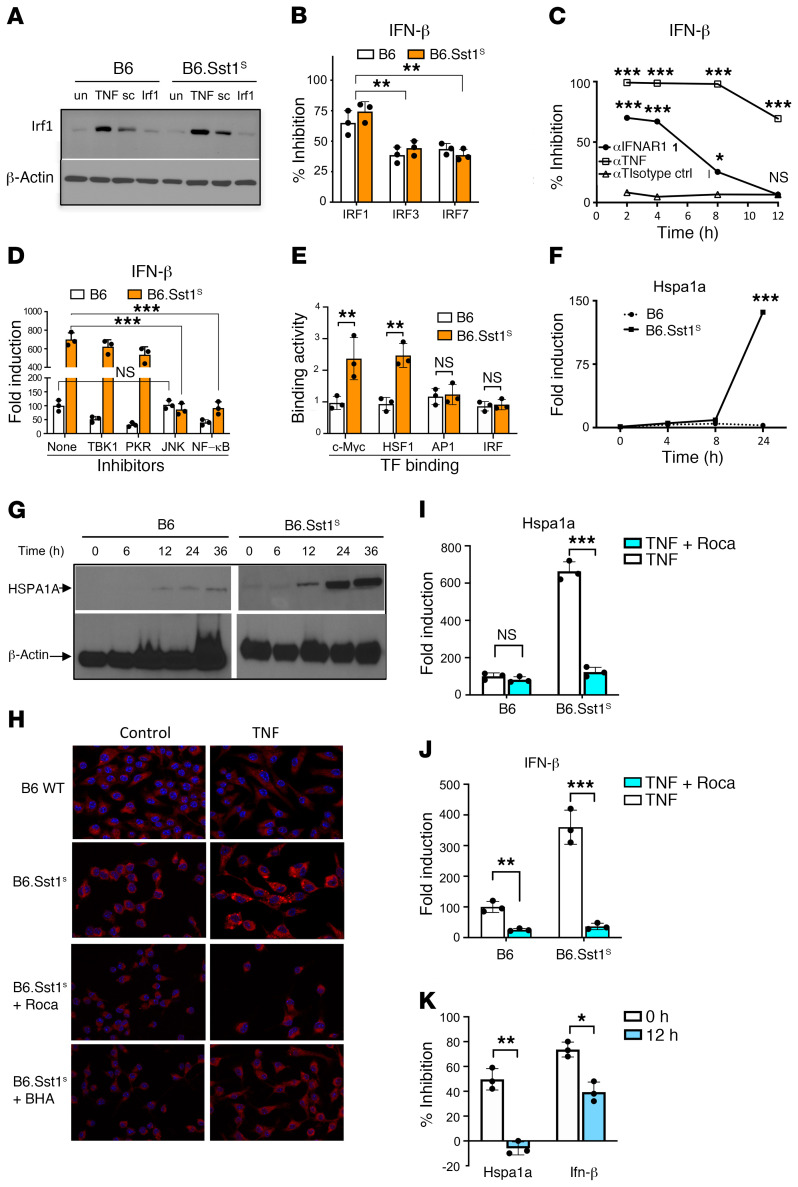
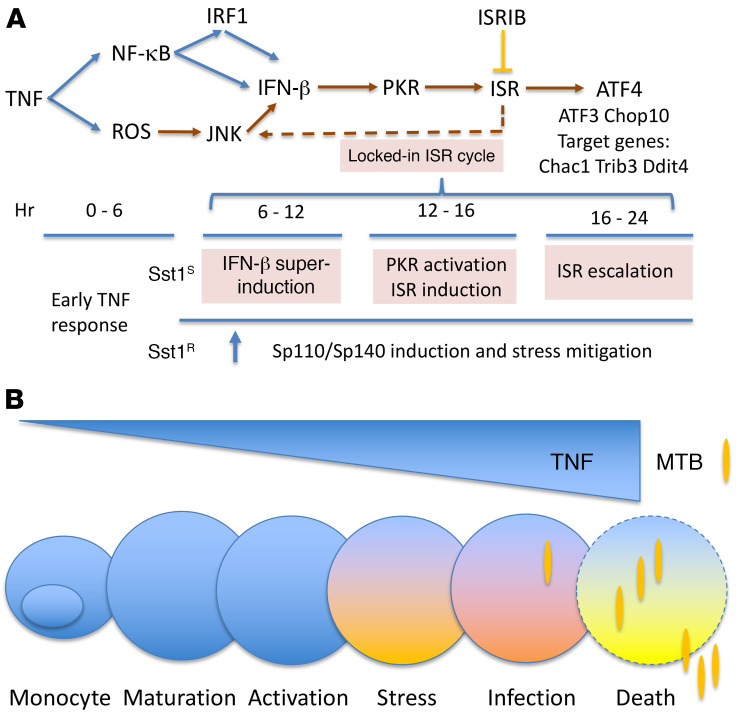
The mechanism by which only some individuals infected with Mycobacterium tuberculosis develop necrotic granulomas with progressive disease while others form controlled granulomas that contain the infection remains poorly defined. Mice carrying the sst1-suscepible (sst1S) genotype develop necrotic inflammatory lung lesions, similar to human tuberculosis (TB) granulomas, which are linked to macrophage dysfunction, while their congenic counterpart (B6) mice do not. In this study we report that (a) sst1S macrophages developed aberrant, biphasic responses to TNF characterized by superinduction of stress and type I interferon pathways after prolonged TNF stimulation; (b) the late-stage TNF response was driven via a JNK/IFN-β/protein kinase R (PKR) circuit; and (c) induced the integrated stress response (ISR) via PKR-mediated eIF2α phosphorylation and the subsequent hyperinduction of ATF3 and ISR-target genes Chac1, Trib3, and Ddit4. The administration of ISRIB, a small-molecule inhibitor of the ISR, blocked the development of necrosis in lung granulomas of M. tuberculosis-infected sst1S mice and concomitantly reduced the bacterial burden. Hence, induction of the ISR and the locked-in state of escalating stress driven by the type I IFN pathway in sst1S macrophages play a causal role in the development of necrosis in TB granulomas. Interruption of the aberrant stress response with inhibitors such as ISRIB may offer novel host-directed therapy strategies.
Keywords: Immunology; Infectious disease; Tuberculosis.
Conflict of interest statement
Figures

References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
