

Comprehensive Structural and Molecular Comparison of Spike Proteins of SARS-CoV-2, SARS-CoV and MERS-CoV, and Their Interactions with ACE2
- PMID: 33302501
- PMCID: PMC7763676
- DOI: 10.3390/cells9122638
Comprehensive Structural and Molecular Comparison of Spike Proteins of SARS-CoV-2, SARS-CoV and MERS-CoV, and Their Interactions with ACE2
Abstract
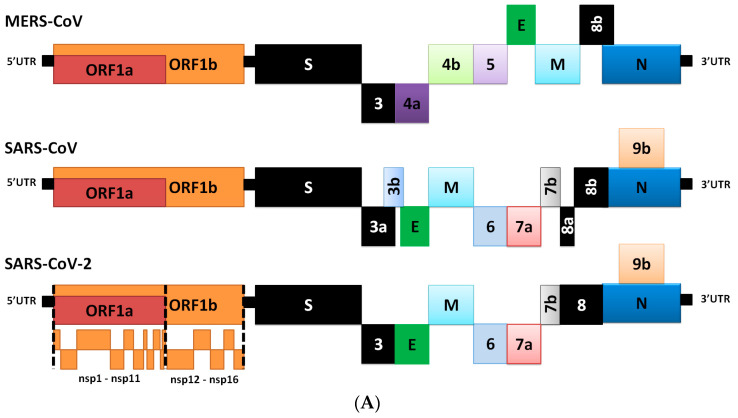
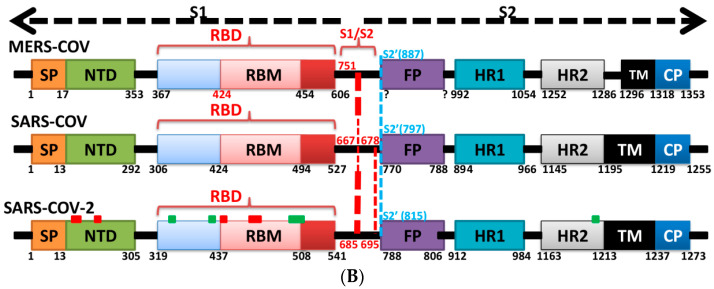
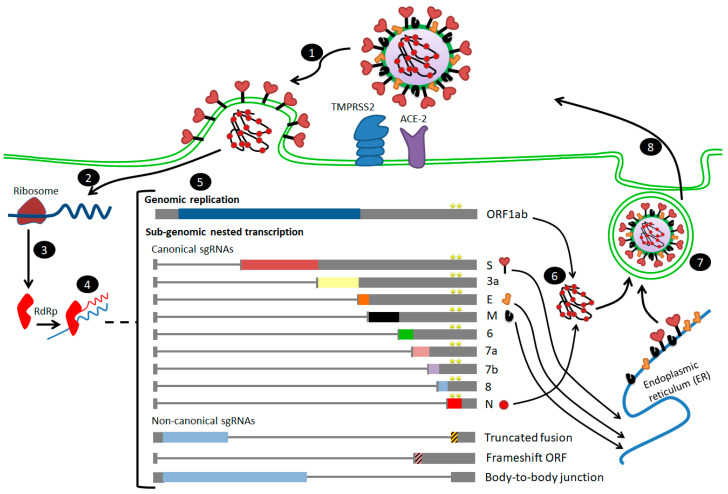
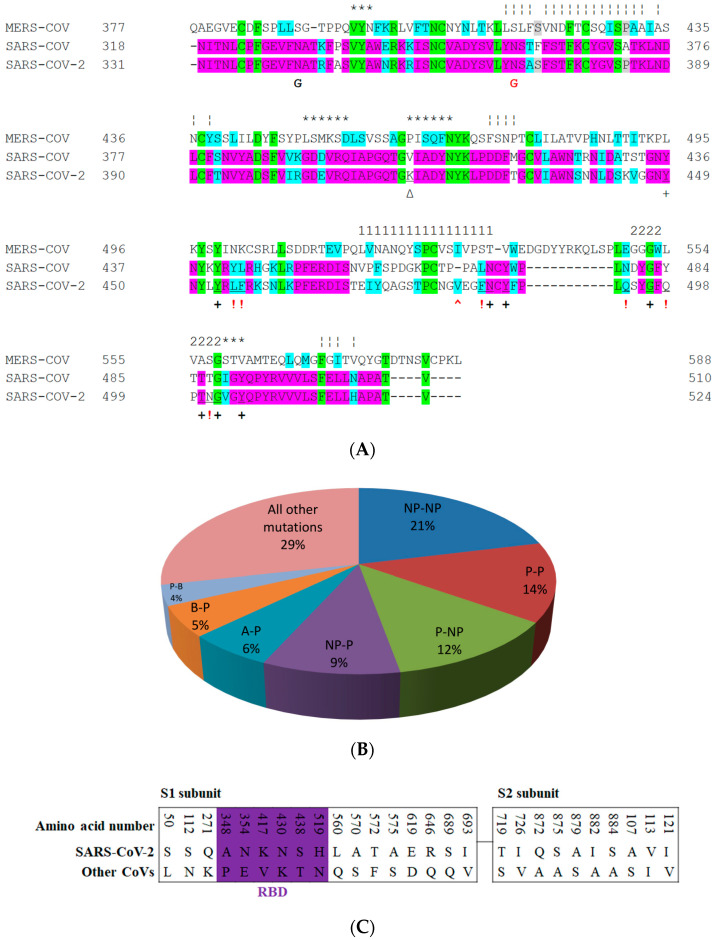
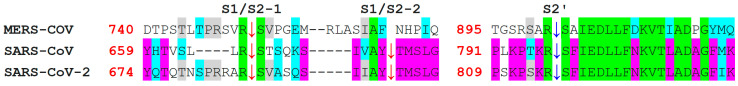
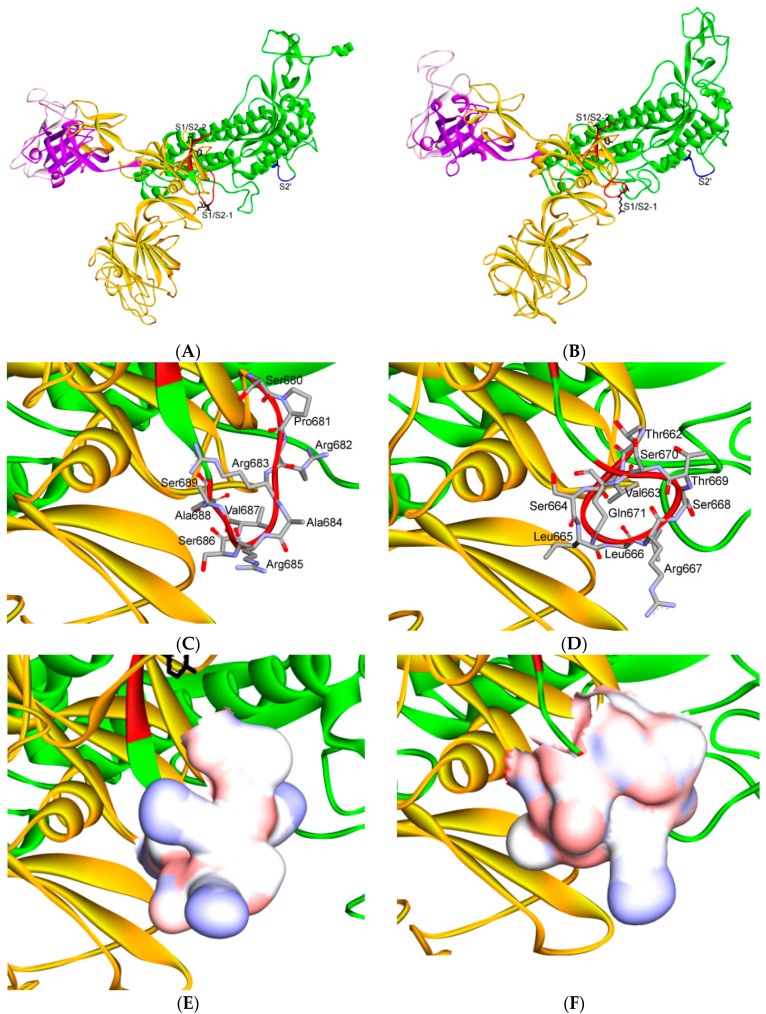
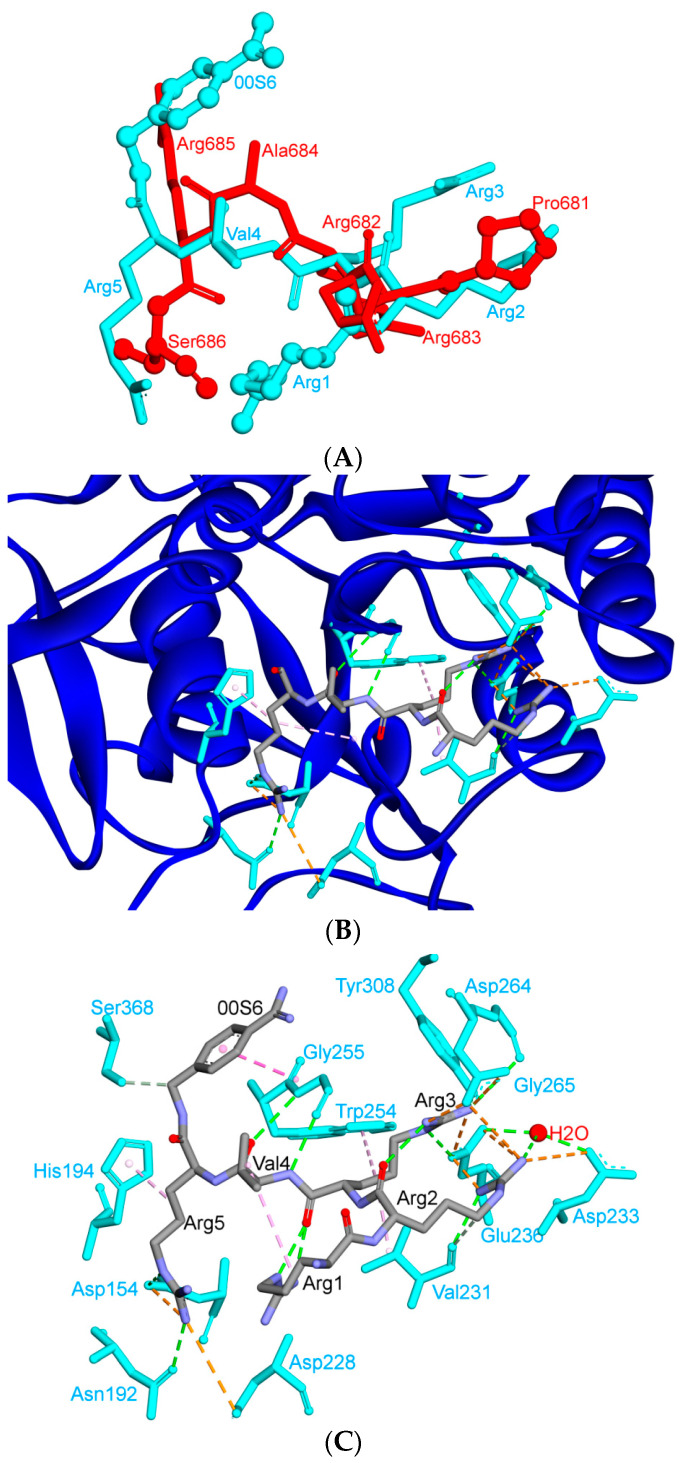
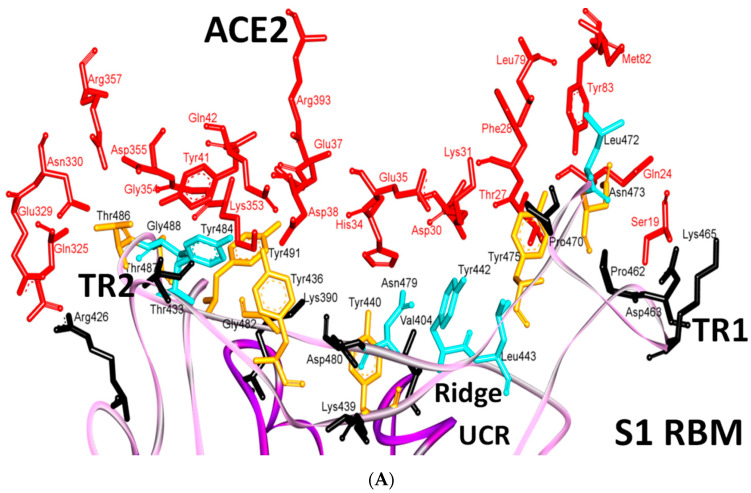
The severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) has recently emerged in China and caused a disease called coronavirus disease 2019 (COVID-19). The virus quickly spread around the world, causing a sustained global outbreak. Although SARS-CoV-2, and other coronaviruses, SARS-CoV and Middle East respiratory syndrome CoV (MERS-CoV) are highly similar genetically and at the protein production level, there are significant differences between them. Research has shown that the structural spike (S) protein plays an important role in the evolution and transmission of SARS-CoV-2. So far, studies have shown that various genes encoding primarily for elements of S protein undergo frequent mutation. We have performed an in-depth review of the literature covering the structural and mutational aspects of S protein in the context of SARS-CoV-2, and compared them with those of SARS-CoV and MERS-CoV. Our analytical approach consisted in an initial genome and transcriptome analysis, followed by primary, secondary and tertiary protein structure analysis. Additionally, we investigated the potential effects of these differences on the S protein binding and interactions to angiotensin-converting enzyme 2 (ACE2), and we established, after extensive analysis of previous research articles, that SARS-CoV-2 and SARS-CoV use different ends/regions in S protein receptor-binding motif (RBM) and different types of interactions for their chief binding with ACE2. These differences may have significant implications on pathogenesis, entry and ability to infect intermediate hosts for these coronaviruses. This review comprehensively addresses in detail the variations in S protein, its receptor-binding characteristics and detailed structural interactions, the process of cleavage involved in priming, as well as other differences between coronaviruses.
Keywords: COVID-19; coronaviruses; proline; receptor-binding motif; spike protein; terminal region.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
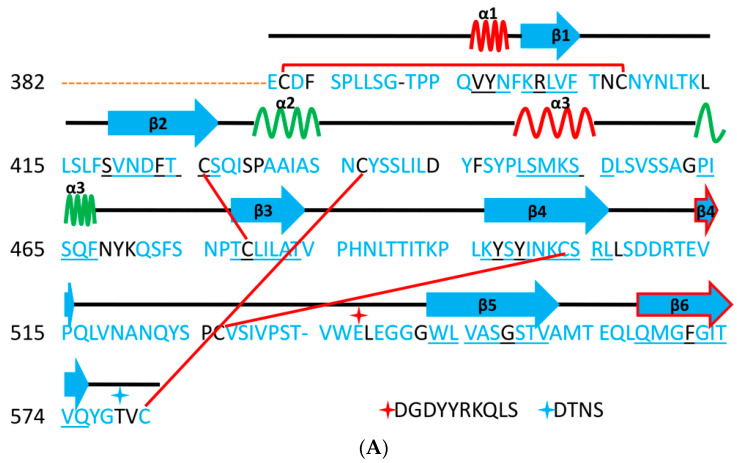
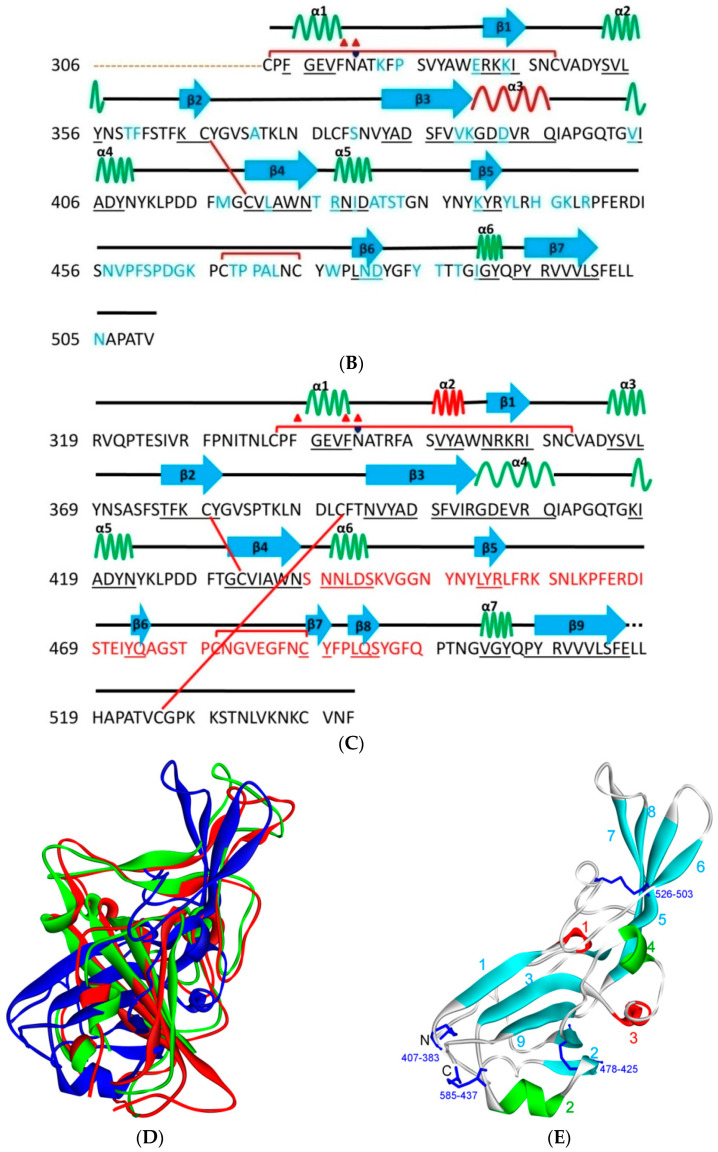
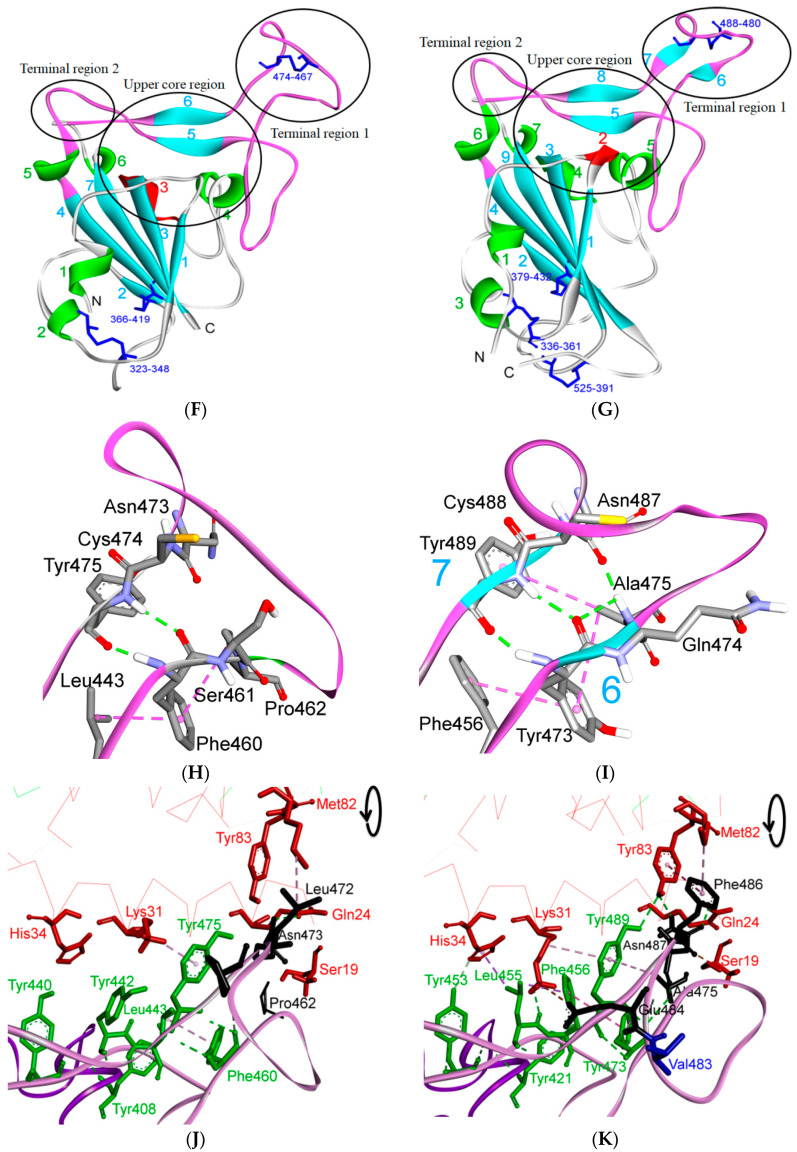
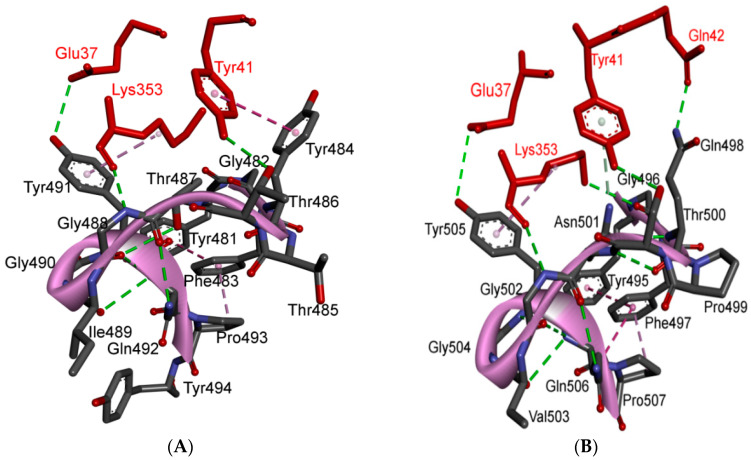
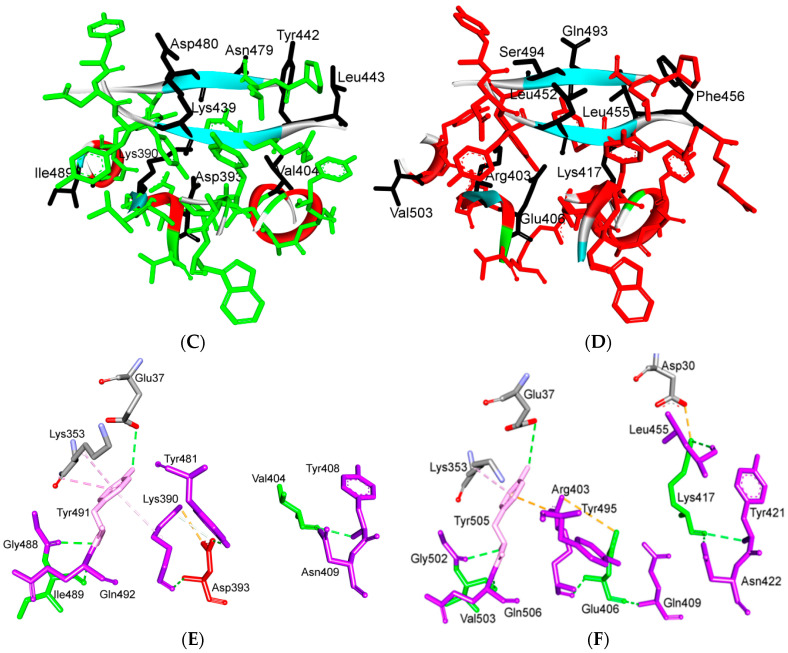
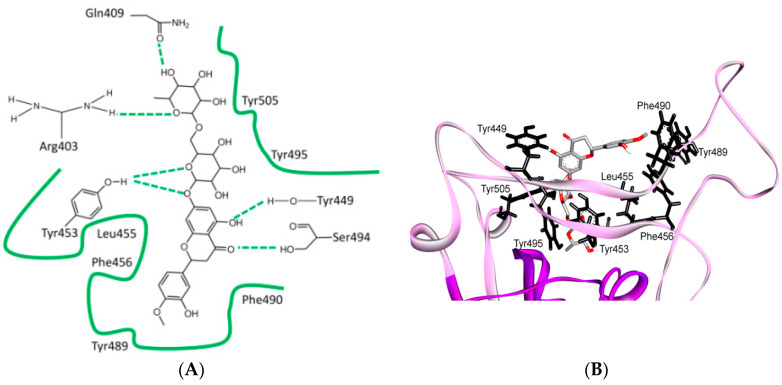
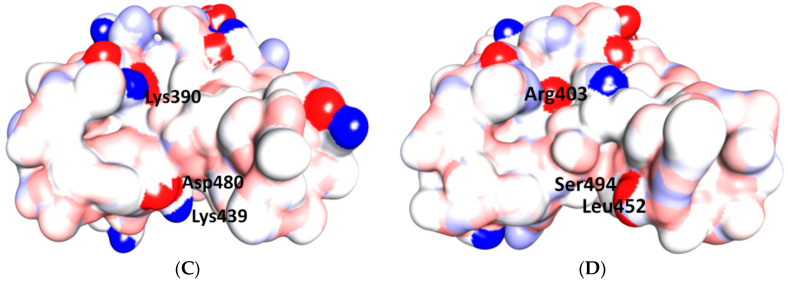
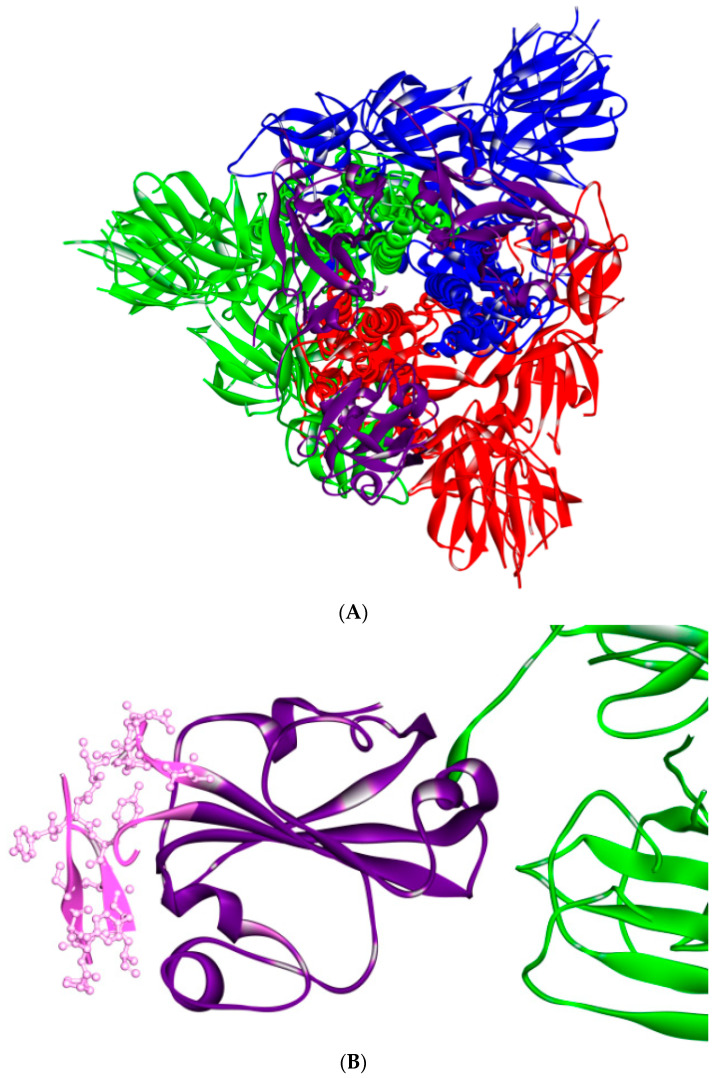
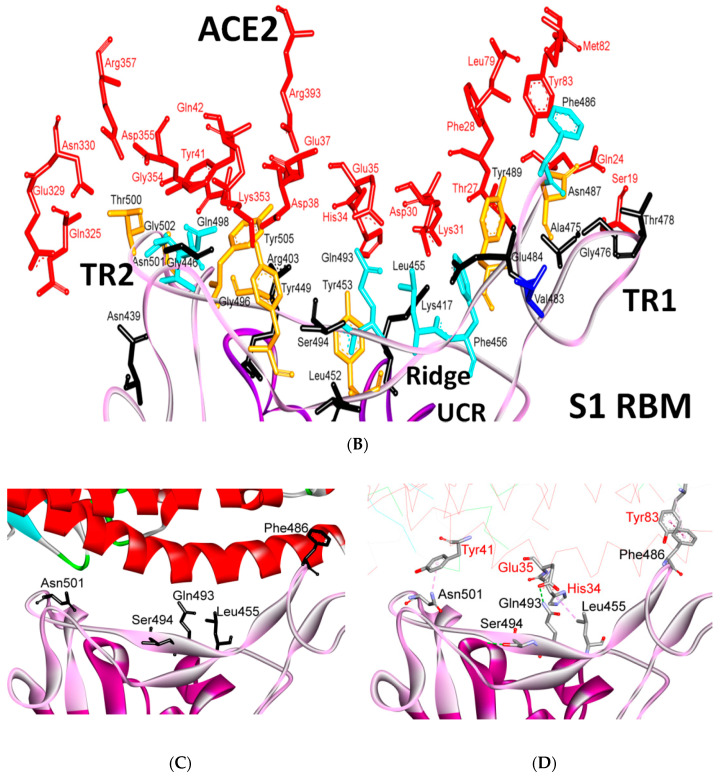
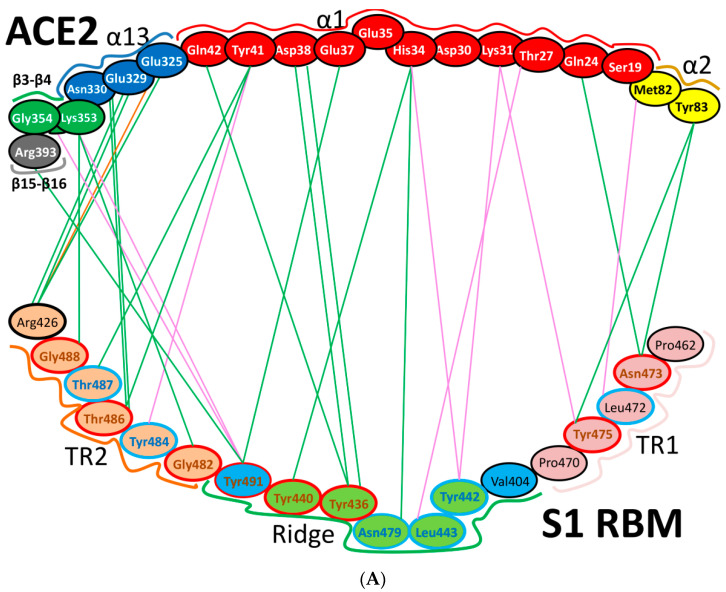
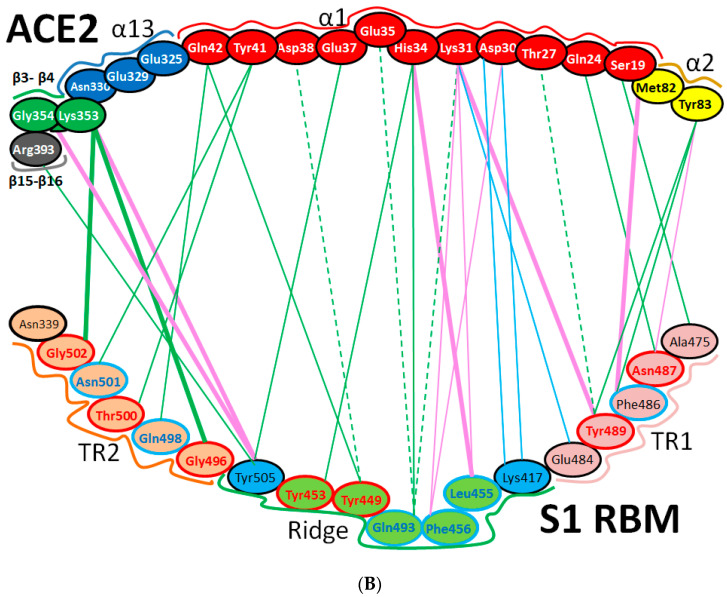
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
