

Hereditary Hypertrophic Cardiomyopathy in Children and Young Adults-The Value of Reevaluating and Expanding Gene Panel Analyses
- PMID: 33302605
- PMCID: PMC7764692
- DOI: 10.3390/genes11121472
Hereditary Hypertrophic Cardiomyopathy in Children and Young Adults-The Value of Reevaluating and Expanding Gene Panel Analyses
Abstract
Introduction: Sudden cardiac death (SCD) and early onset cardiomyopathy (CM) in the young will always lead to suspicion of an underlying genetic disorder. Incited by the rapid advances in genetic testing for disease we have revisited families, which previously tested "gene-negative" for familial predominantly pediatric CM, in hopes of finding a causative gene variant.
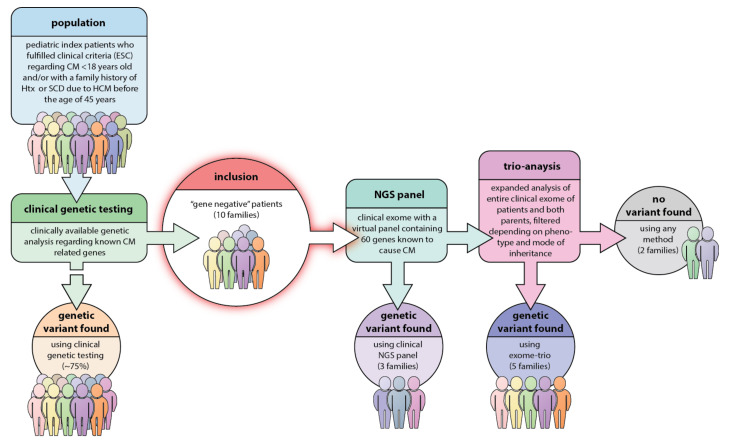
Methods: 10 different families with non-syndromic pediatric CM or hypertrophic cardiomyopathy (HCM) with severe disease progression and/or heredity for HCM/CM related SCD with "gene-negative" results were included. The index patient underwent genetic testing with a recently updated gene panel for CM and SCD. In case of failure to detect a pathogenic variant in a relevant gene, the index patient and both parents underwent clinical (i.e., partial) exome sequencing (trio-exome) in order to catch pathogenic variants linked to the disease in genes that were not included in the CM panel.
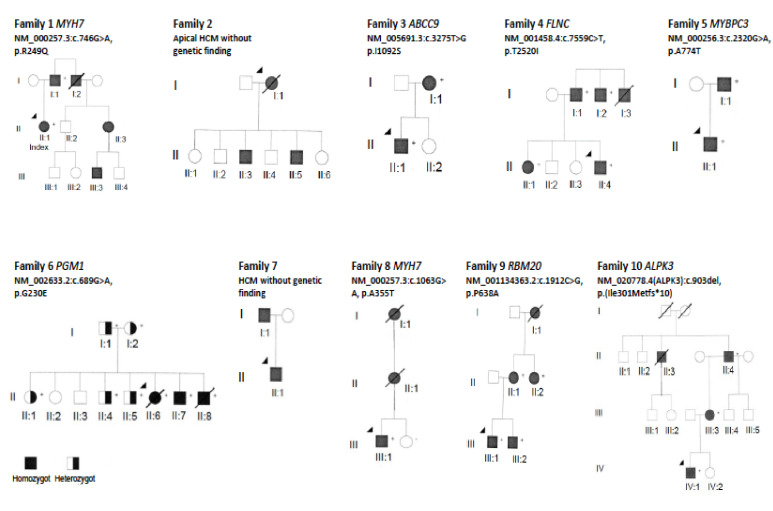
Results: The mean age at clinical presentation of the 10 index cases was 12.5 years (boys 13.4 years, n = 8; girls 9 years, n = 2) and the family history burden was 33 HCM/CM cases including 9 HCM-related SCD and one heart transplantation. In 5 (50%) families we identified a genetic variant classified as pathogenic or likely pathogenic, in accordance with the American College of Medical Genetics and Genomics (ACMG) criteria, in MYH7 (n = 2), RBM20, ALPK3, and PGM1, respectively, and genetic variants of unknown significance (VUS) segregating with the disease in an additional 3 (30%) families, in MYBPC3, ABCC9, and FLNC, respectively.
Conclusion: Our results show the importance of renewed thorough clinical assessment and the necessity to challenge previous genetic test results with more comprehensive updated gene panels or exome sequencing if the initial test failed to identify a causative gene for early onset CM or SCD in children. In pediatric cardiomyopathy cases when the gene panel still fails to detect a causative variant, a trio exome sequencing strategy might resolve some unexplained cases, especially if a multisystemic condition is clinically missed.
Keywords: exome sequencing; gene panel; hypertrophic; pediatric cardiomyopathy; sudden cardiac death.
Conflict of interest statement
The authors declare that they have no conflict of interest or competing interest.
Figures
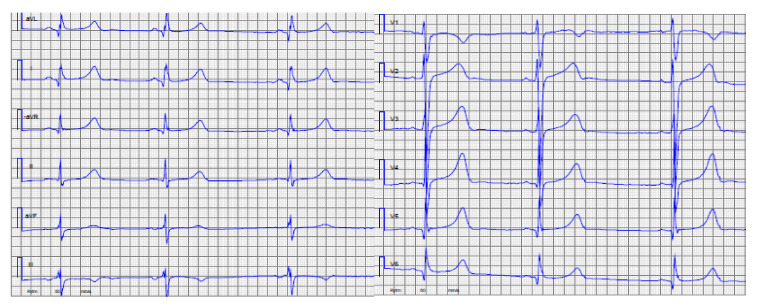
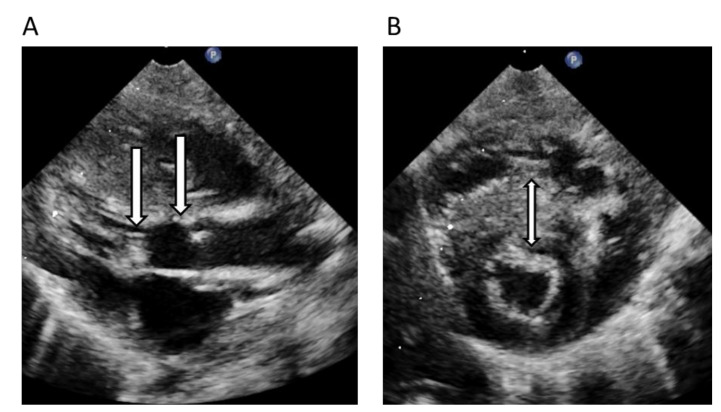
References
-
- Colan S.D., Lipshultz S.E., Lowe A.M., Sleeper L.A., Messere J., Cox G.F., Lurie P.R., Orav E.J., Towbin J.A. Epidemiology and cause-specific outcome of hypertrophic cardiomyopathy in children: Findings from the Pediatric Cardiomyopathy Registry. Circulation. 2007;115:773–781. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
