

Genetic interaction mapping informs integrative structure determination of protein complexes
- PMID: 33303586
- PMCID: PMC7946025
- DOI: 10.1126/science.aaz4910
Genetic interaction mapping informs integrative structure determination of protein complexes
Abstract
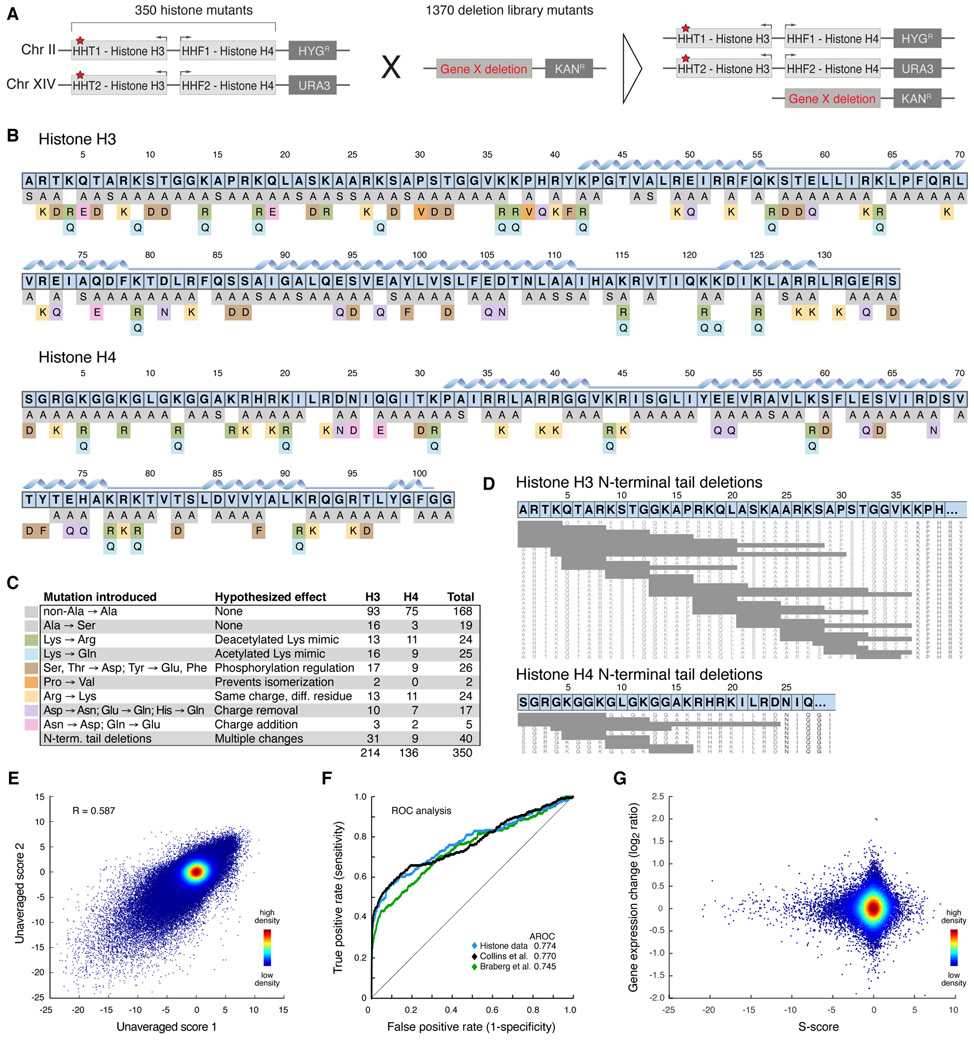
Determining structures of protein complexes is crucial for understanding cellular functions. Here, we describe an integrative structure determination approach that relies on in vivo measurements of genetic interactions. We construct phenotypic profiles for point mutations crossed against gene deletions or exposed to environmental perturbations, followed by converting similarities between two profiles into an upper bound on the distance between the mutated residues. We determine the structure of the yeast histone H3-H4 complex based on ~500,000 genetic interactions of 350 mutants. We then apply the method to subunits Rpb1-Rpb2 of yeast RNA polymerase II and subunits RpoB-RpoC of bacterial RNA polymerase. The accuracy is comparable to that based on chemical cross-links; using restraints from both genetic interactions and cross-links further improves model accuracy and precision. The approach provides an efficient means to augment integrative structure determination with in vivo observations.
Copyright © 2020 The Authors, some rights reserved; exclusive licensee American Association for the Advancement of Science. No claim to original U.S. Government Works.
Figures







Comment in
-
Using genetics to reveal protein structure.Science. 2020 Dec 11;370(6522):1269-1270. doi: 10.1126/science.abf3863. Science. 2020. PMID: 33303603 No abstract available.
References
-
- Alber F, Forster F, Korkin D, Topf M, Sali A, Integrating diverse data for structure determination of macromolecular assemblies. Annu. Rev. Biochem 77, 443–477 (2008). - PubMed
-
- Herzog F et al., Structural probing of a protein phosphatase 2A network by chemical cross-linking and mass spectrometry. Science 337, 1348–1352 (2012). - PubMed
-
- Alber F et al., Determining the architectures of macromolecular assemblies. Nature 450, 683–694 (2007). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- P01 HL089707/HL/NHLBI NIH HHS/United States
- R35 GM127029/GM/NIGMS NIH HHS/United States
- U54 NS100717/NS/NINDS NIH HHS/United States
- U54 RR020839/RR/NCRR NIH HHS/United States
- T32 GM008284/GM/NIGMS NIH HHS/United States
- U54 GM103520/GM/NIGMS NIH HHS/United States
- R01 GM098101/GM/NIGMS NIH HHS/United States
- P01 GM105473/GM/NIGMS NIH HHS/United States
- P50 GM082250/GM/NIGMS NIH HHS/United States
- U54 CA209891/CA/NCI NIH HHS/United States
- P50 AI150476/AI/NIAID NIH HHS/United States
- R01 GM084279/GM/NIGMS NIH HHS/United States
- R35 GM118061/GM/NIGMS NIH HHS/United States
- R01 GM083960/GM/NIGMS NIH HHS/United States
- U19 AI135990/AI/NIAID NIH HHS/United States
- P50 GM081879/GM/NIGMS NIH HHS/United States
- R01 GM123159/GM/NIGMS NIH HHS/United States
- R01 GM084448/GM/NIGMS NIH HHS/United States
- DP5 OD009180/OD/NIH HHS/United States
- R35 GM126900/GM/NIGMS NIH HHS/United States
- P41 GM109824/GM/NIGMS NIH HHS/United States
- S10 OD021596/OD/NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
