

Genomic recombination events may reveal the evolution of coronavirus and the origin of SARS-CoV-2
- PMID: 33303849
- PMCID: PMC7728743
- DOI: 10.1038/s41598-020-78703-6
Genomic recombination events may reveal the evolution of coronavirus and the origin of SARS-CoV-2
Erratum in
-
Author Correction: Genomic recombination events may reveal the evolution of coronavirus and the origin of SARS-CoV-2.Sci Rep. 2021 Nov 23;11(1):23077. doi: 10.1038/s41598-021-02549-9. Sci Rep. 2021. PMID: 34815545 Free PMC article. No abstract available.
Abstract
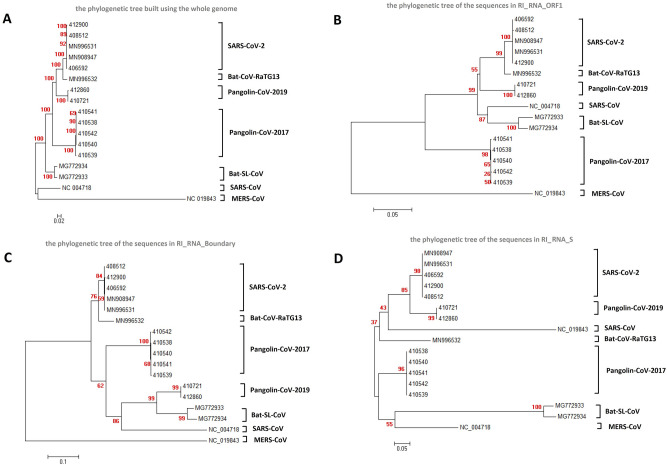
To trace the evolution of coronaviruses and reveal the possible origin of the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), which causes the coronavirus disease 2019 (COVID-19), we collected and thoroughly analyzed 29,452 publicly available coronavirus genomes, including 26,312 genomes of SARS-CoV-2 strains. We observed coronavirus recombination events among different hosts including 3 independent recombination events with statistical significance between some isolates from humans, bats and pangolins. Consistent with previous records, we also detected putative recombination between strains similar or related to Bat-CoV-RaTG13 and Pangolin-CoV-2019. The putative recombination region is located inside the receptor-binding domain (RBD) of the spike glycoprotein (S protein), which may represent the origin of SARS-CoV-2. Population genetic analyses provide estimates suggesting that the putative introduced DNA within the RBD is undergoing directional evolution. This may result in the adaptation of the virus to hosts. Unsurprisingly, we found that the putative recombination region in S protein was highly diverse among strains from bats. Bats harbor numerous coronavirus subclades that frequently participate in recombination events with human coronavirus. Therefore, bats may provide a pool of genetic diversity for the origin of SARS-CoV-2.
Conflict of interest statement
The authors declare no competing interests.
Figures


References
Publication types
MeSH terms
Grants and funding
- 31200941/National Natural Science Foundation of China/International
- 106112016CDJXY290002/Fundamental Research Funds for the Central Universities/International
- 2019YFC1604600/National Key Research and Development Program/International
- 19226631D/National Natural Science Foundation of HeBei province/International
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
