

E6-mediated activation of JNK drives EGFR signalling to promote proliferation and viral oncoprotein expression in cervical cancer
- PMID: 33303976
- PMCID: PMC8166842
- DOI: 10.1038/s41418-020-00693-9
E6-mediated activation of JNK drives EGFR signalling to promote proliferation and viral oncoprotein expression in cervical cancer
Abstract
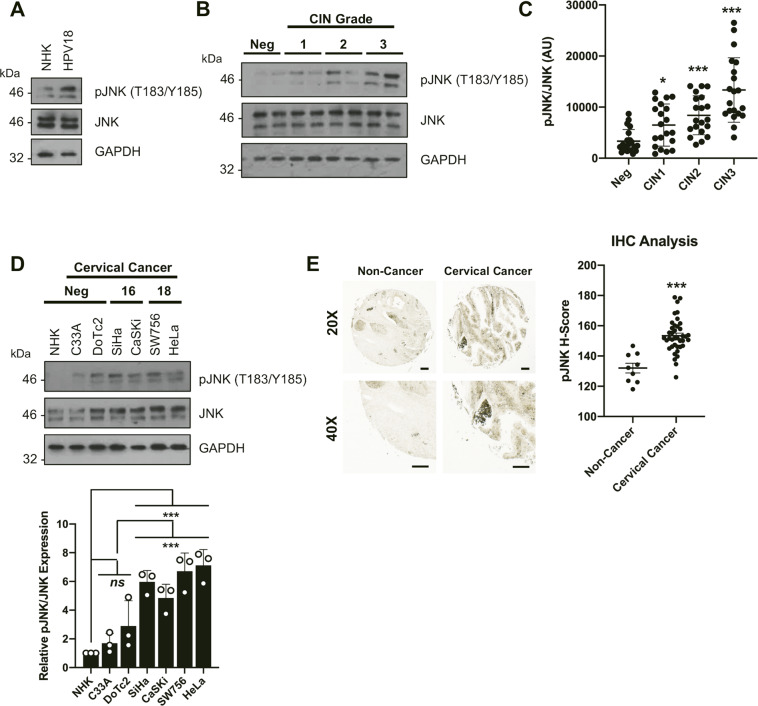
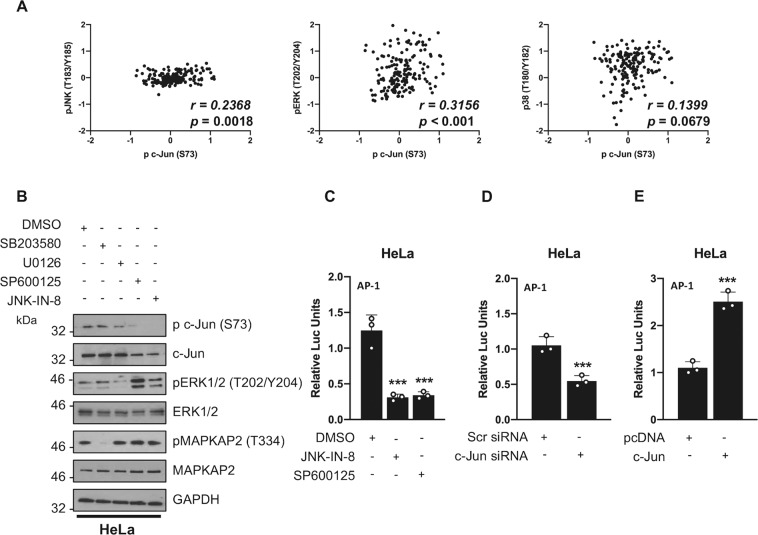
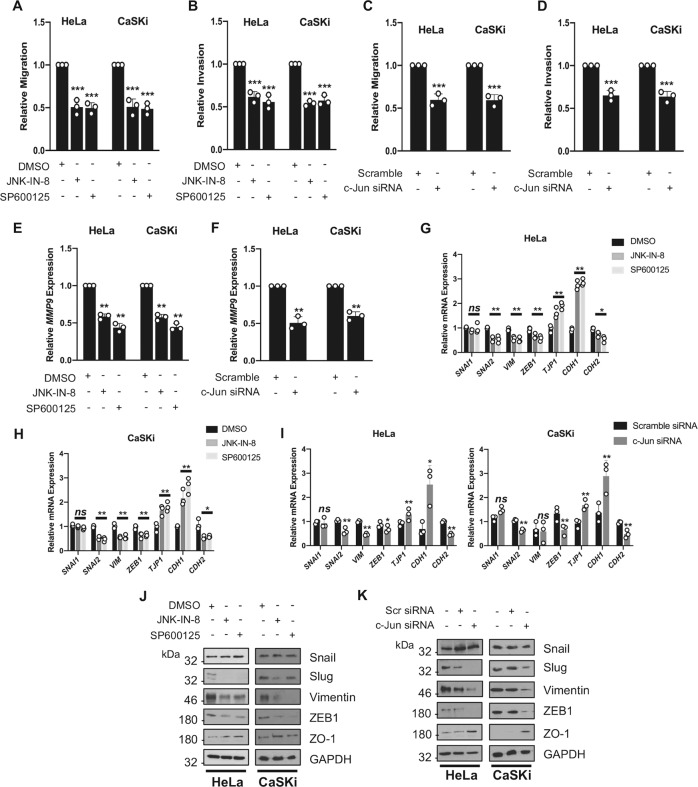
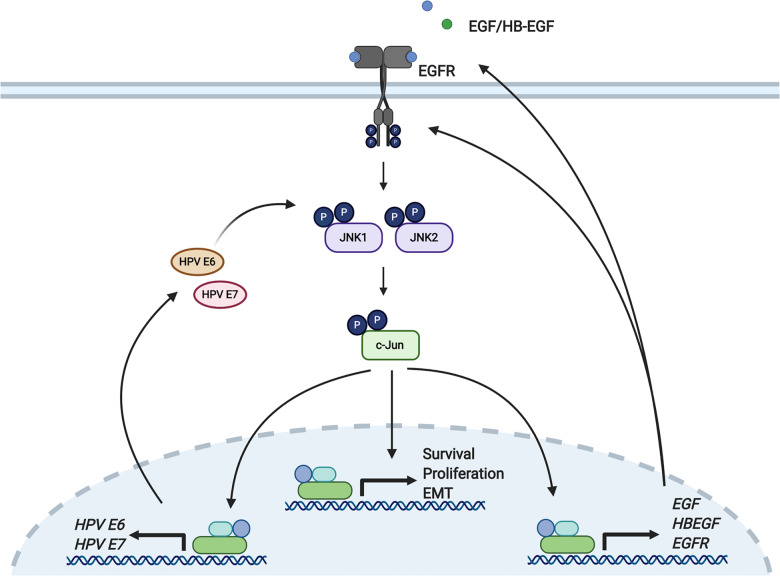
Human papillomaviruses (HPV) are a major cause of malignancy worldwide, contributing to ~5% of all human cancers including almost all cases of cervical cancer and a growing number of ano-genital and oral cancers. HPV-induced malignancy is primarily driven by the viral oncogenes, E6 and E7, which manipulate host cellular pathways to increase cell proliferation and enhance cell survival, ultimately predisposing infected cells to malignant transformation. Consequently, a more detailed understanding of viral-host interactions in HPV-associated disease offers the potential to identify novel therapeutic targets. Here, we identify that the c-Jun N-terminal kinase (JNK) signalling pathway is activated in cervical disease and in cervical cancer. The HPV E6 oncogene induces JNK1/2 phosphorylation in a manner that requires the E6 PDZ binding motif. We show that blockade of JNK1/2 signalling using small molecule inhibitors, or knockdown of the canonical JNK substrate c-Jun, reduces cell proliferation and induces apoptosis in cervical cancer cells. We further demonstrate that this phenotype is at least partially driven by JNK-dependent activation of EGFR signalling via increased expression of EGFR and the EGFR ligands EGF and HB-EGF. JNK/c-Jun signalling promoted the invasive potential of cervical cancer cells and was required for the expression of the epithelial to mesenchymal transition (EMT)-associated transcription factor Slug and the mesenchymal marker Vimentin. Furthermore, JNK/c-Jun signalling is required for the constitutive expression of HPV E6 and E7, which are essential for cervical cancer cell growth and survival. Together, these data demonstrate a positive feedback loop between the EGFR signalling pathway and HPV E6/E7 expression, identifying a regulatory mechanism in which HPV drives EGFR signalling to promote proliferation, survival and EMT. Thus, our study has identified a novel therapeutic target that may be beneficial for the treatment of cervical cancer.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures




References
-
- Hausen zur H. Papillomaviruses and cancer: from basic studies to clinical application. Nat Rev Cancer. 2002;2:342–50. - PubMed
-
- Crosbie EJ, Einstein MH, Franceschi S, Kitchener HC. Human papillomavirus and cervical cancer. Lancet. 2013;382:889–99. - PubMed
-
- Wasson CW, Morgan EL, Müller M, Ross RL, Hartley M, Roberts S, et al. Human papillomavirus type 18 E5 oncogene supports cell cycle progression and impairs epithelial differentiation by modulating growth factor receptor signalling during the virus life cycle. Oncotarget. 2017;8:103581–600. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
