

SHP2 Inhibition Influences Therapeutic Response to Tepotinib in Tumors with MET Alterations
- PMID: 33305187
- PMCID: PMC7718487
- DOI: 10.1016/j.isci.2020.101832
SHP2 Inhibition Influences Therapeutic Response to Tepotinib in Tumors with MET Alterations
Abstract
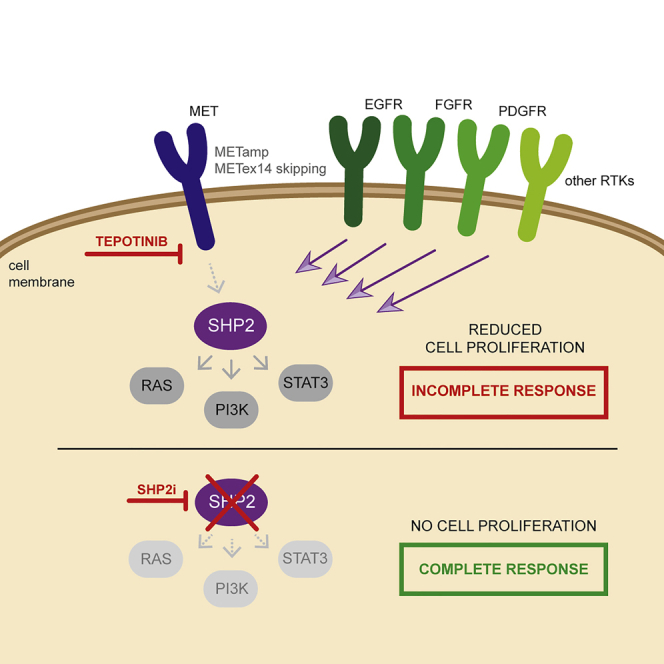
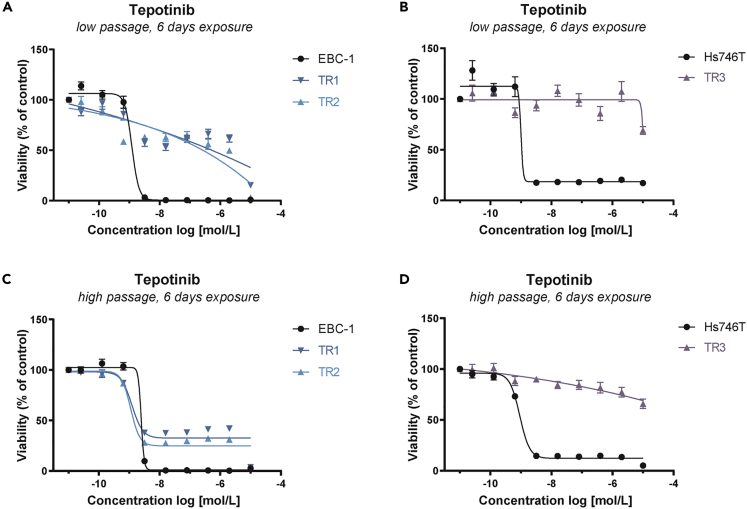
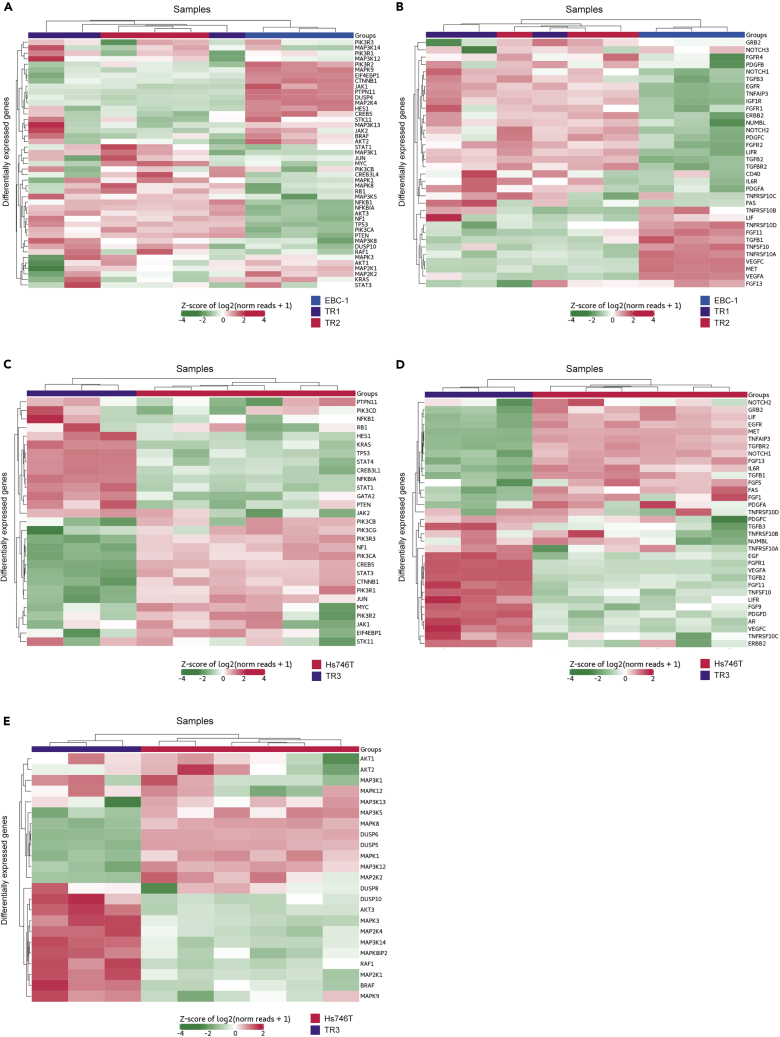
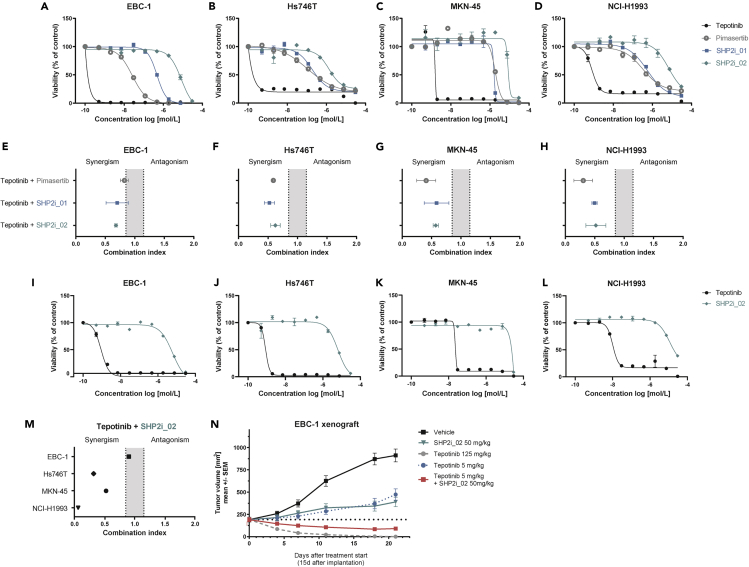
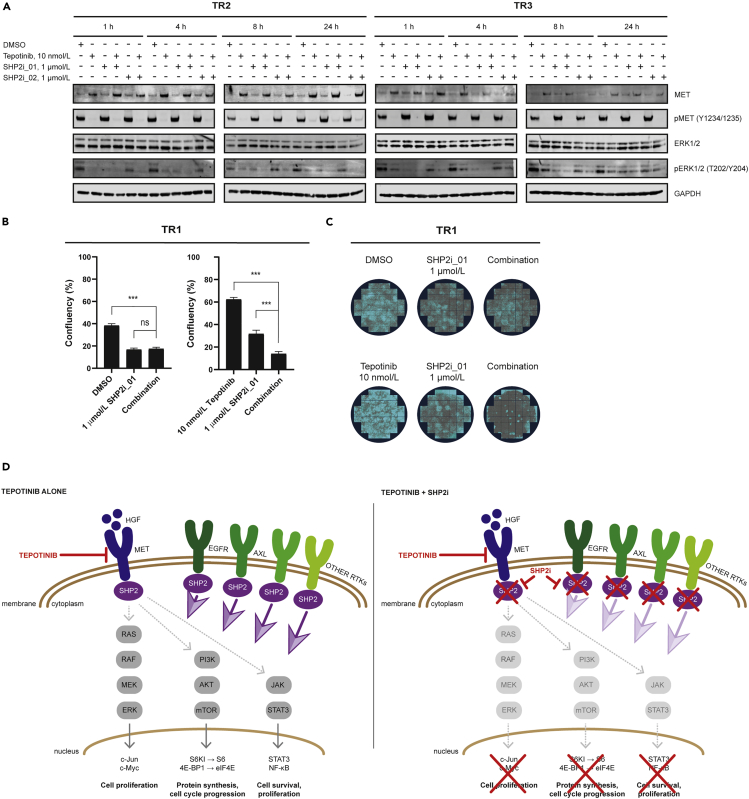
Tepotinib is an oral MET inhibitor approved for metastatic non-small cell lung cancer (NSCLC) harboring MET exon 14 (METex14) skipping mutations. Examining treatment-naive or tepotinib-resistant cells with MET amplification or METex14 skipping mutations identifies other receptor tyrosine kinases (RTKs) that co-exist in cells prior to tepotinib exposure and become more prominent upon tepotinib resistance. In a small cohort of patients with lung cancer with MET genetic alterations treated with tepotinib, gene copy number gains of other RTKs were found at baseline and affected treatment outcome. An Src homology 2 domain-containing phosphatase 2 (SHP2) inhibitor delayed the emergence of tepotinib resistance and synergized with tepotinib in treatment-naive and tepotinib-resistant cells as well as in xenograft models. Alternative signaling pathways potentially diminish the effect of tepotinib monotherapy, and the combination of tepotinib with an SHP2 inhibitor enables the control of tumor growth in cells with MET genetic alterations.
Keywords: Cancer; Treatment.
© 2020 The Author(s).
Conflict of interest statement
L.P., F.J., C.R., C.S., N.L., M.S., D.M., M.K., C.E., A.B., K.M.S., and N.K. are employees of Merck KGaA. A.B. and M.S. hold stocks in Merck KGaA. No other potential conflict of interest relevant to this article was reported.
Figures



References
-
- Awad M.M., Oxnard G.R., Jackman D.M., Savukoski D.O., Hall D., Shivdasani P., Heng J.C., Dahlberg S.E., Janne P.A., Verma S. MET exon 14 mutations in non-small-cell lung cancer are associated with advanced age and stage-dependent MET genomic amplification and c-met overexpression. J. Clin. Oncol. 2016;34:721–730. - PubMed
-
- Backes A., Zech B., Felber B., Klebl B., Muller G. Small-molecule inhibitors binding to protein kinase. Part II: the novel pharmacophore approach of type II and type III inhibition. Expert Opin. Drug Discov. 2008;3:1427–1449. - PubMed
LinkOut - more resources
Full Text Sources
Miscellaneous
