

Late-Onset Aicardi-Goutières Syndrome: A Characterization of Presenting Clinical Features
- PMID: 33307271
- PMCID: PMC7856674
- DOI: 10.1016/j.pediatrneurol.2020.10.012
Late-Onset Aicardi-Goutières Syndrome: A Characterization of Presenting Clinical Features
Abstract
Background: Aicardi-Goutières syndrome (AGS) is a genetic interferonopathy characterized by early onset of severe neurological injury with intracranial calcifications, leukoencephalopathy, and systemic inflammation. Increasingly, a spectrum of neurological dysfunction and presentation beyond the infantile period is being recognized in AGS. The aim of this study was to characterize late-infantile and juvenile-onset AGS.
Methods: We conducted a multi-institution review of individuals with AGS who were older than one year at the time of presentation, including medical history, imaging characteristics, and suspected diagnoses at presentation.
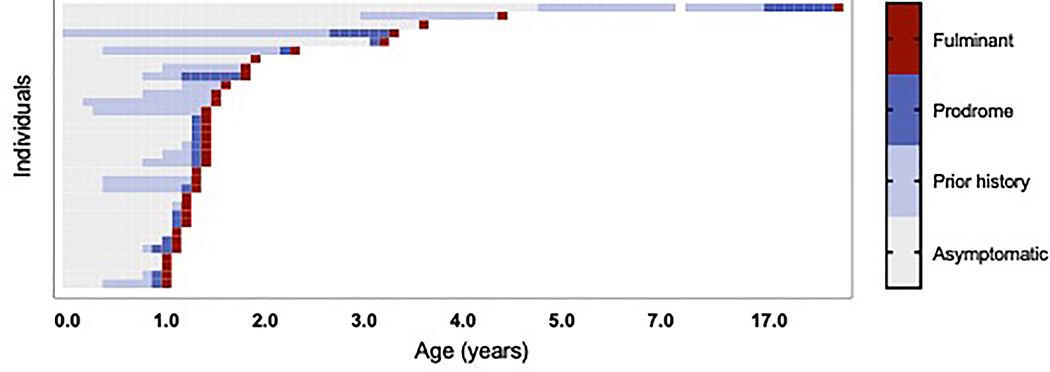
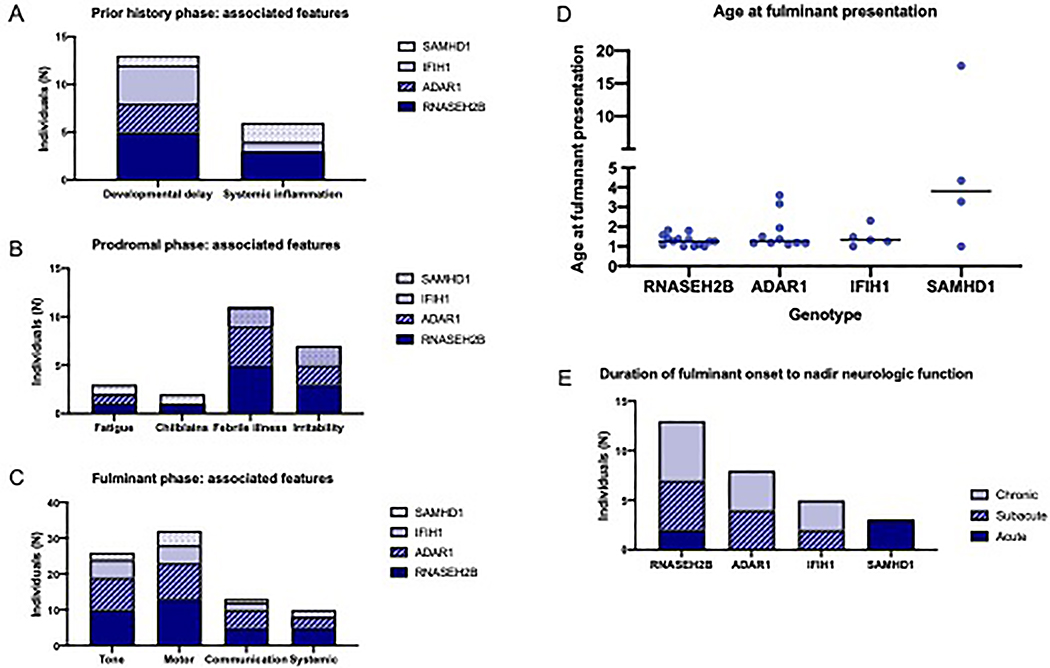
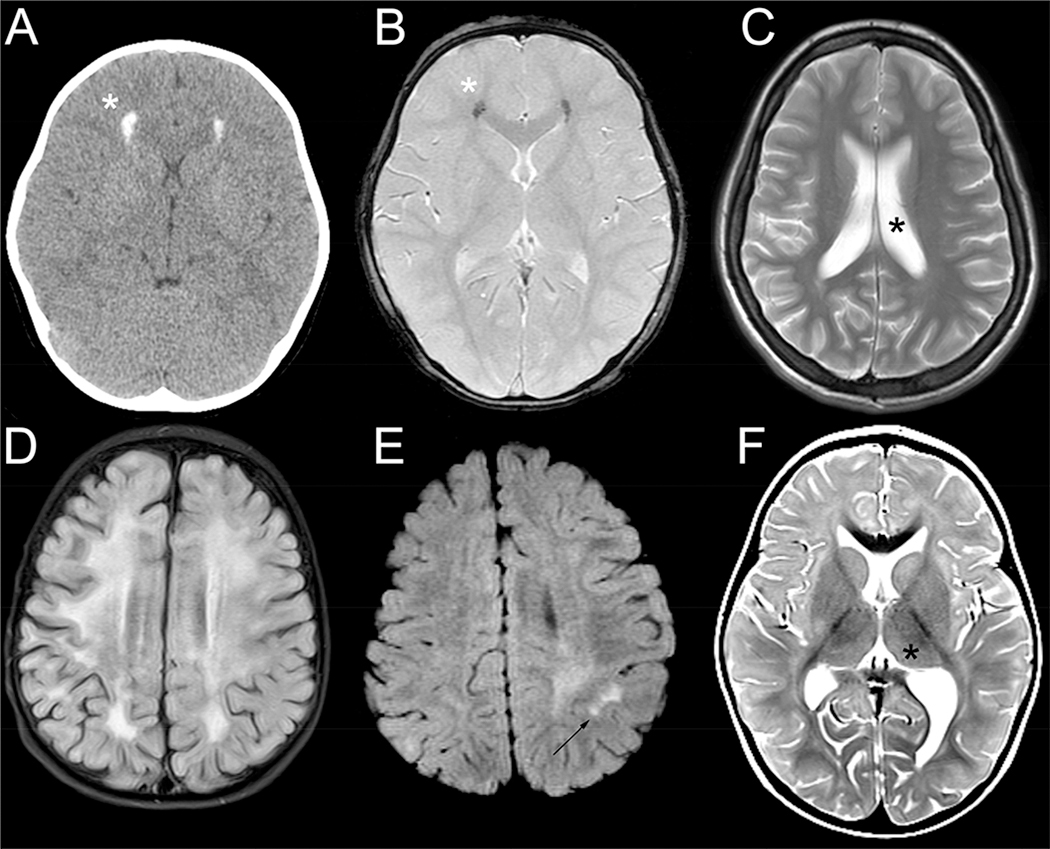
Results: Thirty-four individuals were identified, all with pathogenic variants in RNASEH2B, SAMHD1, ADAR1, or IFIH1. Most individuals had a history of developmental delay and/or systemic symptoms, such as sterile pyrexias and chilblains, followed by a prodromal period associated with increasing symptoms. This was followed by an abrupt onset of neurological decline (fulminant phase), with a median onset at 1.33 years (range 1.00 to 17.68 years). Most individuals presented with a change in gross motor skills (97.0%), typically with increased tone (78.8%). Leukodystrophy was the most common magnetic resonance imaging finding (40.0%). Calcifications were less common (12.9%).
Conclusions: This is the first study to characterize the presentation of late-infantile and juvenile onset AGS and its phenotypic spectrum. Late-onset AGS can present insidiously and lacks classical clinical and neuroimaging findings. Signs of early systemic dysfunction before fulminant disease onset and loss of motor symptoms were common. We strongly recommend genetic testing when there is concern for sustained inflammation of unknown origins or changes in motor skills in children older than one year.
Keywords: ADAR1; Aicardi-Goutières syndrome; IFIH1; Leukodystrophy; RNASEH2B; SAMHD1; Type I interferonopathy.
Copyright © 2020 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Figures
References
-
- Livingston JH and Crow YJ, Neurologic Phenotypes Associated with Mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, ADAR1, and IFIH1: Aicardi-Goutieres Syndrome and Beyond. Neuropediatrics, 2016. 47(6): p. 355–360. - PubMed
-
- Aicardi J and Goutieres F, A progressive familial encephalopathy in infancy with calcifications of the basal ganglia and chronic cerebrospinal fluid lymphocytosis. Ann Neurol, 1984. 15(1): p. 49–54. - PubMed
Publication types
MeSH terms
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
