

Heterogeneous Host-Pathogen Encounters Coordinate Antibiotic Resilience in Mycobacterium tuberculosis
- PMID: 33309526
- PMCID: PMC7611257
- DOI: 10.1016/j.tim.2020.10.013
Heterogeneous Host-Pathogen Encounters Coordinate Antibiotic Resilience in Mycobacterium tuberculosis
Abstract
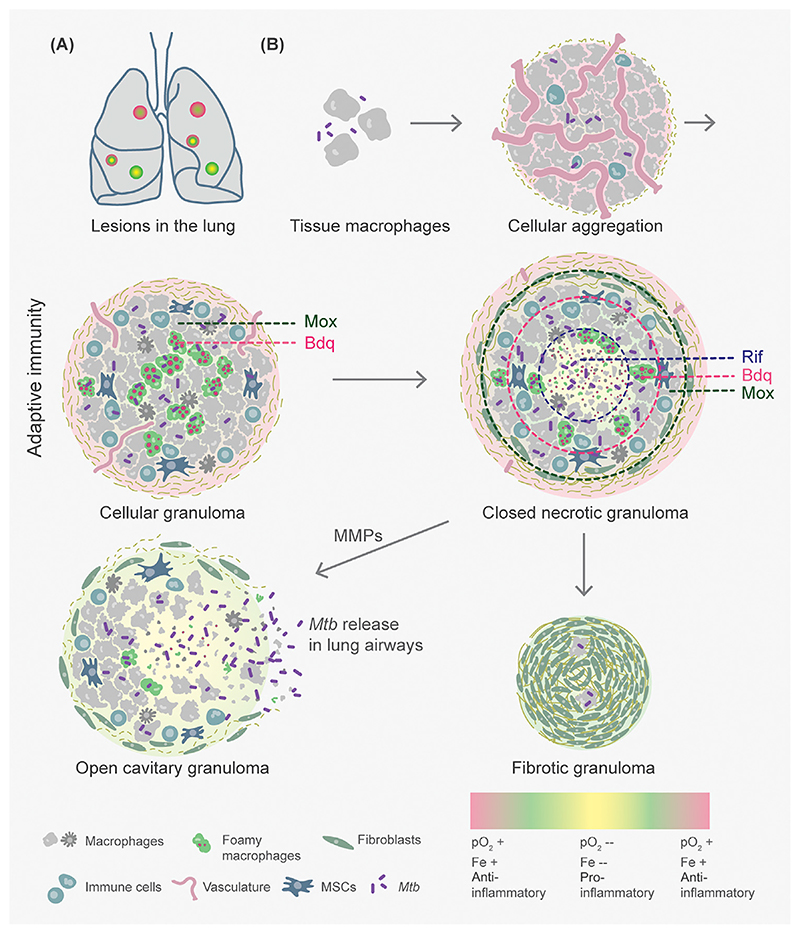
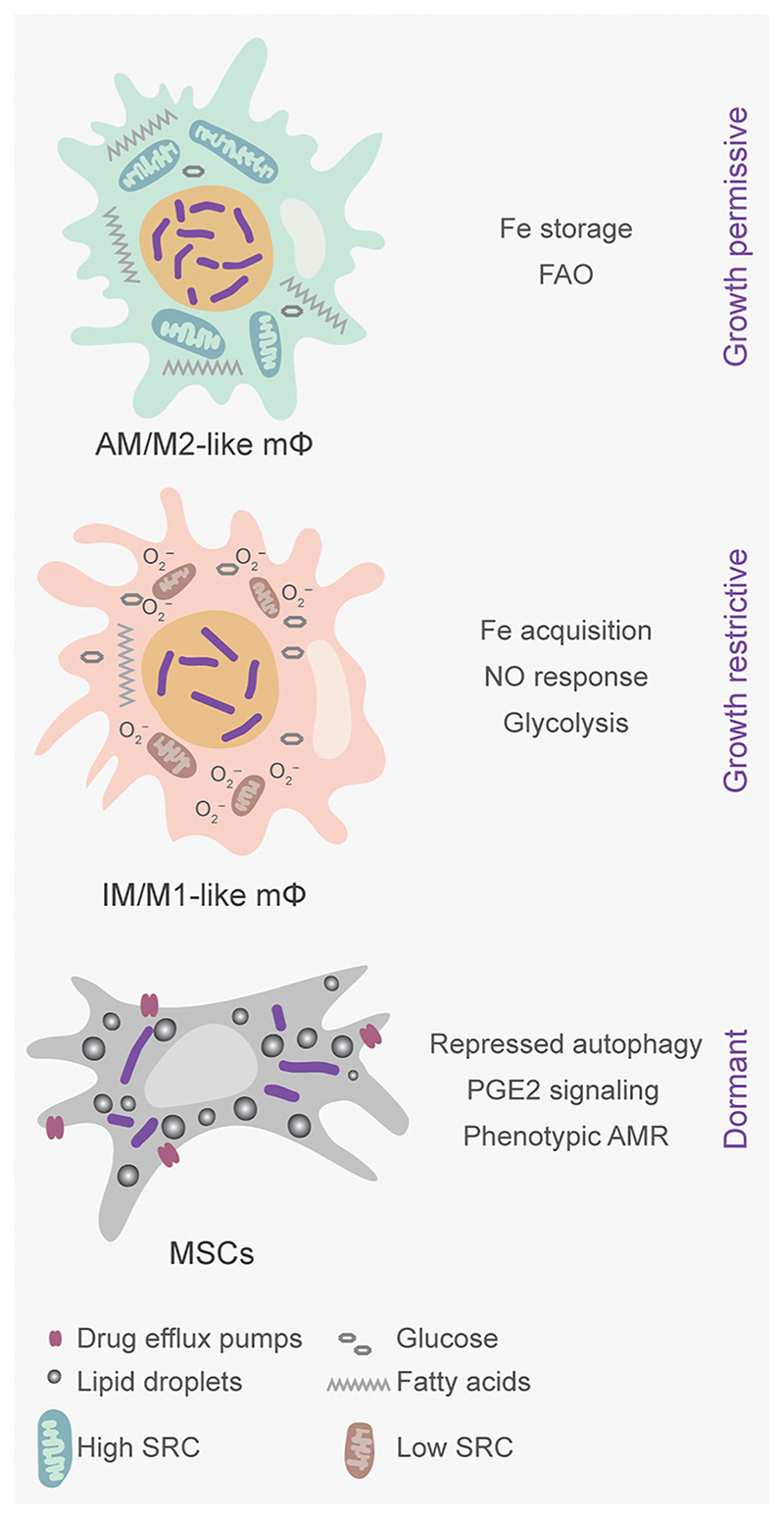
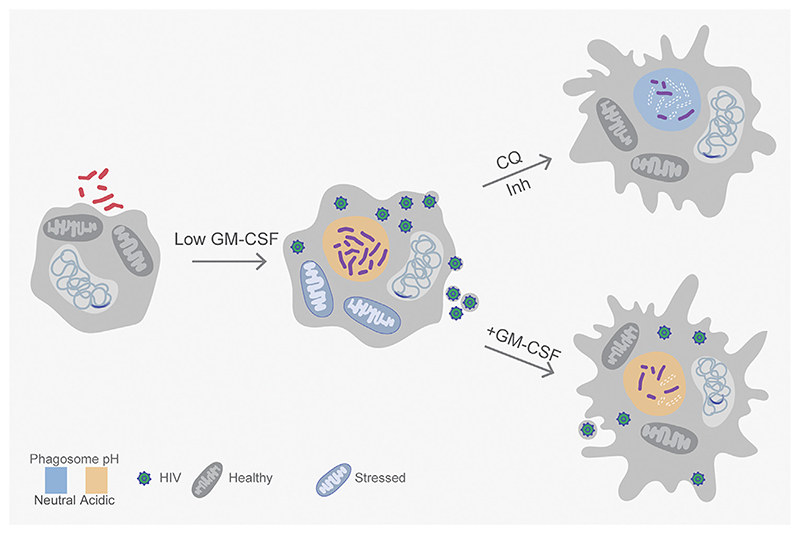
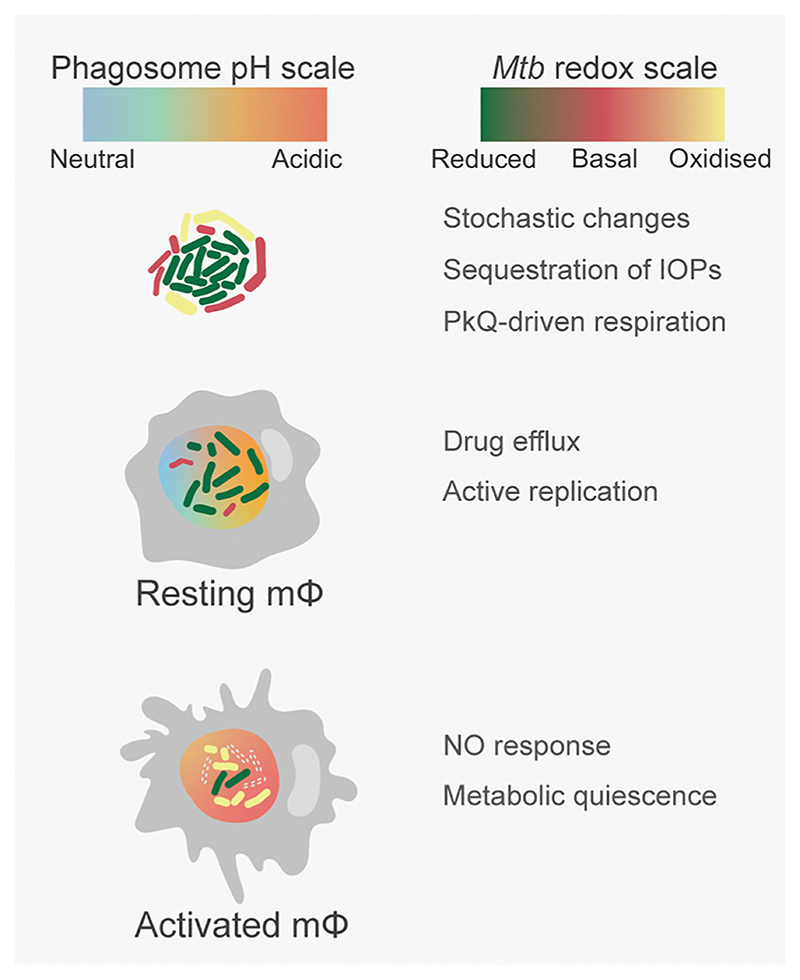
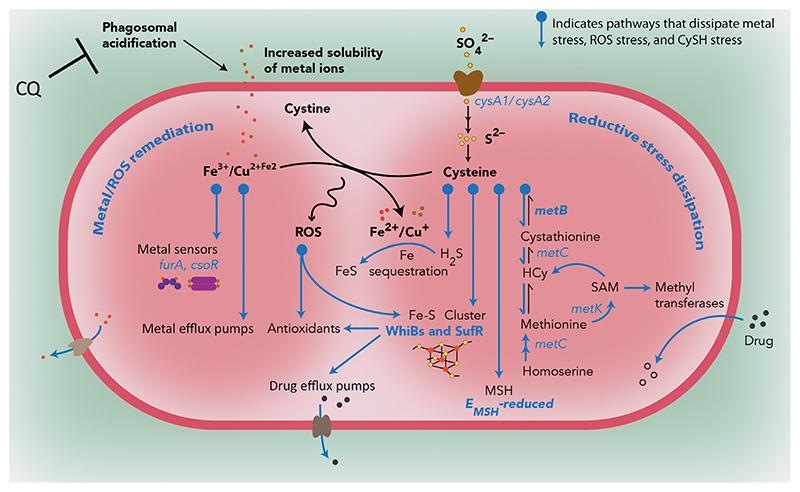
Successful treatment of tuberculosis (TB) depends on the eradication of its causative agent Mycobacterium tuberculosis (Mtb) in the host. However, the emergence of phenotypically drug-resistant Mtb in the host environment tempers the ability of antibiotics to cure disease. Host immunity produces diverse microenvironmental niches that are exploited by Mtb to mobilize adaptation programs. Such differential interactions amplify pre-existing heterogeneity in the host-pathogen milieu to influence disease pathology and therapy outcome. Therefore, comprehending the intricacies of phenotypic heterogeneity can be an empirical step forward in potentiating drug action. With this goal, we review the interconnectedness of the lesional, cellular, and bacterial heterogeneity underlying phenotypic drug resistance. Based on this information, we anticipate the development of new therapeutic strategies targeting host-pathogen heterogeneity to cure TB.
Keywords: drug tolerance; phagosomal acidification; phenotypic heterogeneity; redox metabolism.
Copyright © 2020 Elsevier Ltd. All rights reserved.
Figures
References
-
- Hobby GL, et al. Observations on the mechanism of action of penicillin. Proc Soc Exp Biol Med. 1942;50:281–285.
-
- Bigger JW. Treatment of staphylococcal infections with penicillin by intermittent sterilisation. Lancet. 1944;244:497–500.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
