

Genomic epidemiology reveals transmission patterns and dynamics of SARS-CoV-2 in Aotearoa New Zealand
- PMID: 33311501
- PMCID: PMC7733492
- DOI: 10.1038/s41467-020-20235-8
Genomic epidemiology reveals transmission patterns and dynamics of SARS-CoV-2 in Aotearoa New Zealand
Abstract
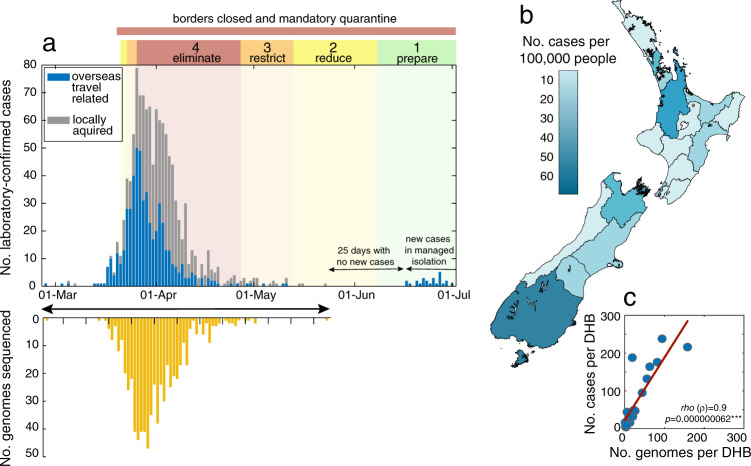
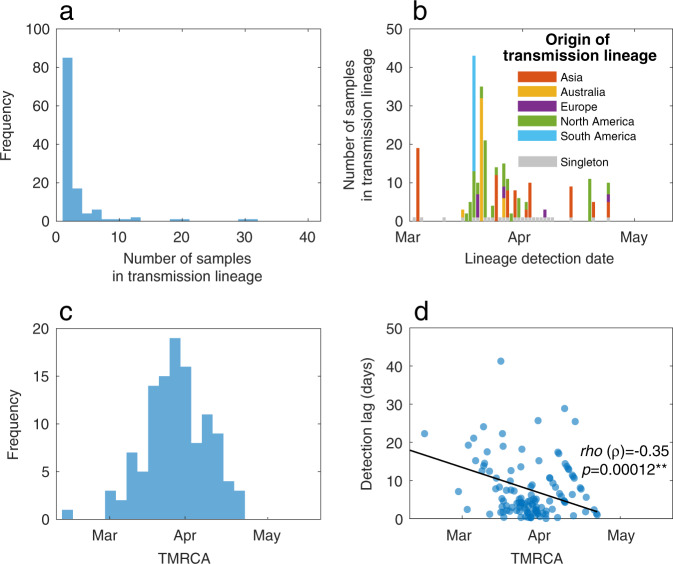
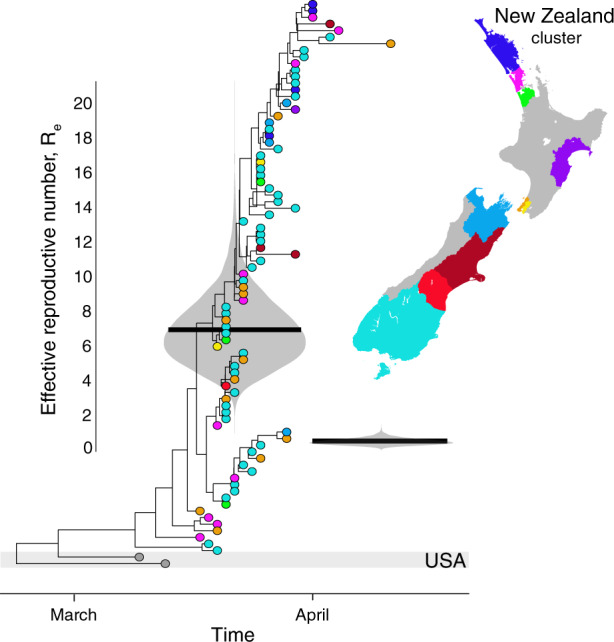
New Zealand, a geographically remote Pacific island with easily sealable borders, implemented a nationwide 'lockdown' of all non-essential services to curb the spread of COVID-19. Here, we generate 649 SARS-CoV-2 genome sequences from infected patients in New Zealand with samples collected during the 'first wave', representing 56% of all confirmed cases in this time period. Despite its remoteness, the viruses imported into New Zealand represented nearly all of the genomic diversity sequenced from the global virus population. These data helped to quantify the effectiveness of public health interventions. For example, the effective reproductive number, Re of New Zealand's largest cluster decreased from 7 to 0.2 within the first week of lockdown. Similarly, only 19% of virus introductions into New Zealand resulted in ongoing transmission of more than one additional case. Overall, these results demonstrate the utility of genomic pathogen surveillance to inform public health and disease mitigation.
Conflict of interest statement
The authors declare no competing interests.
Figures

References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
