

Outcomes of Childhood Cholestasis in Alagille Syndrome: Results of a Multicenter Observational Study
- PMID: 33313463
- PMCID: PMC7049675
- DOI: 10.1002/hep4.1468
Outcomes of Childhood Cholestasis in Alagille Syndrome: Results of a Multicenter Observational Study
Abstract
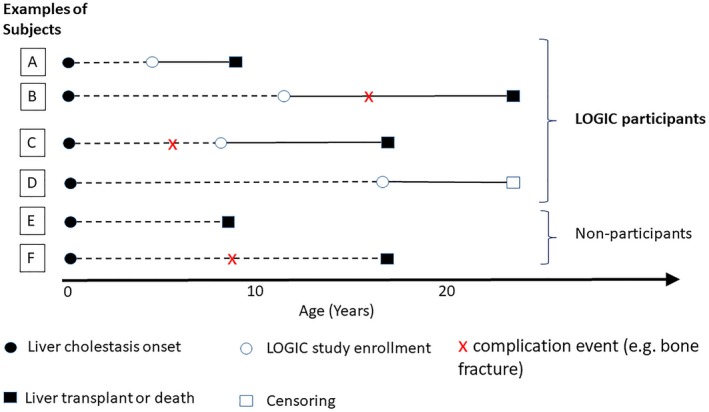
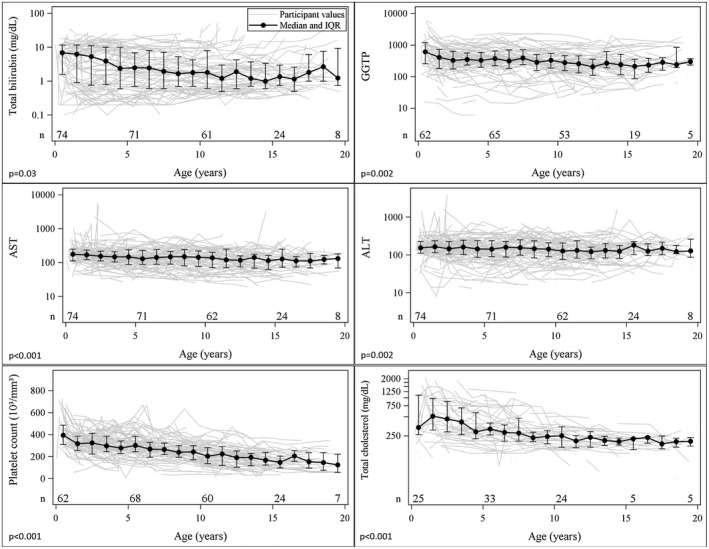
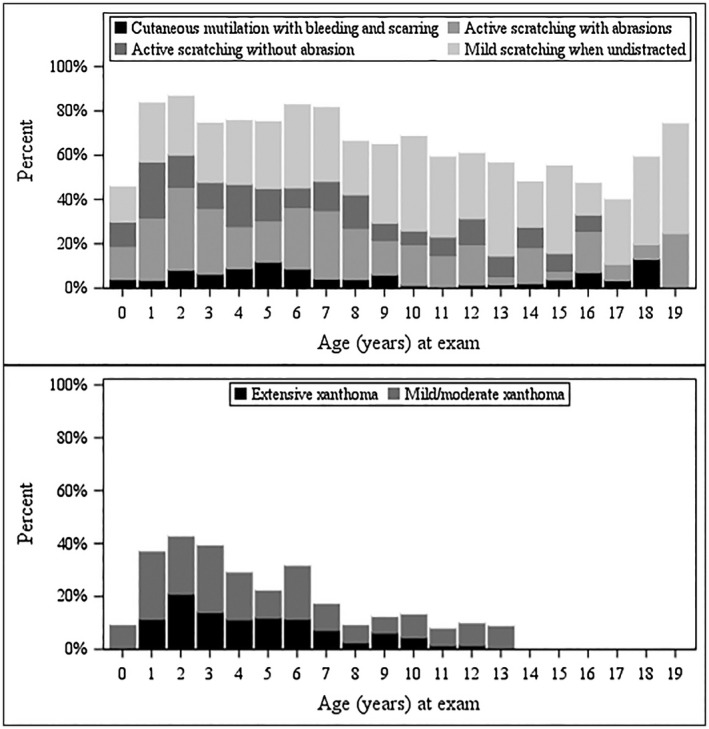
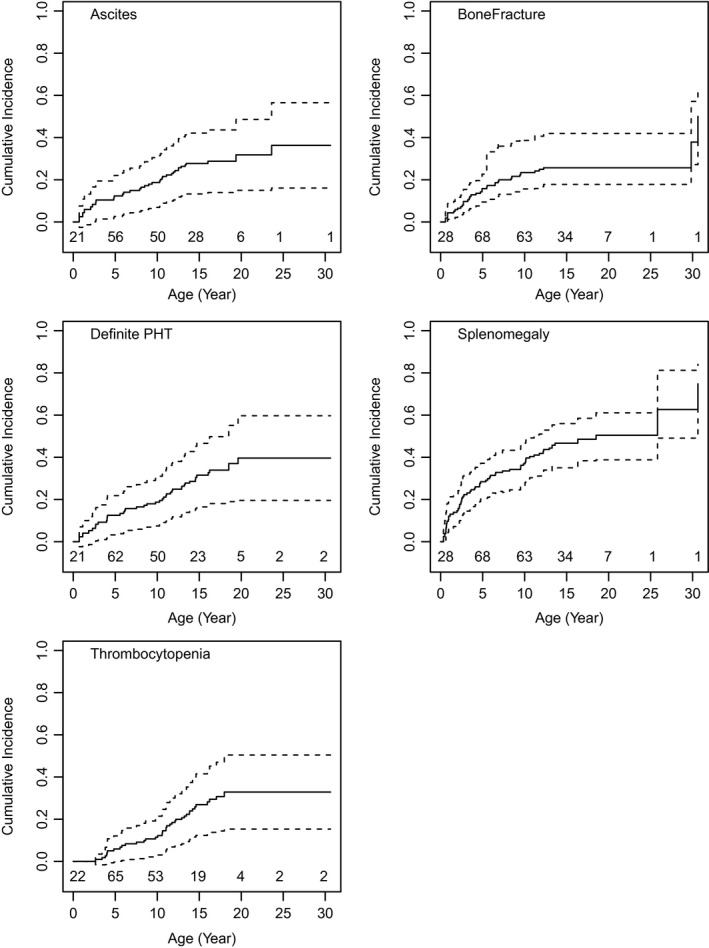
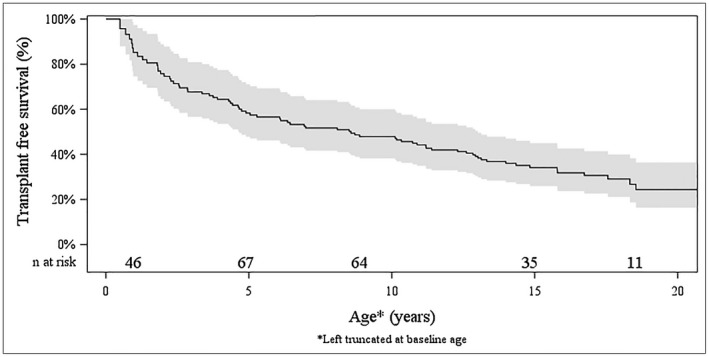
Alagille syndrome (ALGS) is an autosomal dominant multisystem disorder with cholestasis as a defining clinical feature. We sought to characterize hepatic outcomes in a molecularly defined cohort of children with ALGS-related cholestasis. Two hundred and ninety-three participants with ALGS with native liver were enrolled. Participants entered the study at different ages and data were collected retrospectively prior to enrollment, and prospectively during the study course. Genetic analysis in 206 revealed JAGGED1 mutations in 91% and NOTCH2 mutations in 4%. Growth was impaired with mean height and weight z-scores of <-1.0 at all ages. Regression analysis revealed that every 10 mg/dL increase in total bilirubin was associated with a decrease in height z-score by 0.10 (P = 0.03) and weight z-score by 0.15 (P = 0.007). Total bilirubin was higher for younger participants (P = 0.03) with a median of 6.9 mg/dL for those less than 1 year old compared with a median of 1.3 mg/dL for participants 13 years or older. The median gamma glutamyl transferase also dropped from 612 to 268 in the same age groups. After adjusting for age, there was substantial within-individual variation of alanine aminotransferase. By 20 years of age, 40% of participants had developed definite portal hypertension. Estimated liver transplant-free survival at the age of 18.5 years was 24%. Conclusions: This is the largest multicenter natural history study of cholestasis in ALGS, demonstrating a previously underappreciated burden of liver disease with early profound cholestasis, a second wave of portal hypertension later in childhood, and less than 25% of patients reaching young adulthood with their native liver. These findings will promote optimization of ALGS management and development of clinically relevant endpoints for future therapeutic trials.
© 2020 The Authors. Hepatology Communications published by Wiley Periodicals, Inc., on behalf of the American Association for the Study of Liver Diseases.
Conflict of interest statement
Potential conflict of interest: Dr. Thompson consults for, advises, received grants from, and owns stock in Generation Bio and Qing Bile Therapeutics. He consults for, advises, and received grants from Albireo and Mirum. He consults for and advises Alnylam, Horizon, and Sana. Dr. Rosenthal consults for and received grants from Gilead, AbbVie, and Retrophin. He consults for Albireo, Mirum, and Audentes. He received grants from BMS and Merck. Dr. Heubi consults for and is on the speakers’ bureau for Retrophin. He consults for and received grants from Mirum. He advises Alnylam. He received grants from Friesland Campina. He has equity interest in Asklepion. Dr. Karpen consults for Albireo, Intercept, and Mirum. Dr. Loomes consults for Mirum and Albireo. Dr. Mack consults for Albireo. Dr. Kamath consults and received grants from Mirum. She consults for Albireo and DCI. Dr. Leung consults for Merck. He received grants from AbbVie and Gilead. Dr. Molleston received grants from Shire, Mirum, Gilead, and AbbVie. Dr. Murray received grants from Gilead. Dr. Sokol consults for Retrophin, Shire, Mirum, and Albireo. Dr. Romero reports research grants from Merck and Gilead. Dr. Spinner consults for Retrophin. All other authors have nothing to report.
Figures
References
-
- Alagille D. Alagille syndrome today. Clin Invest Med 1996;19:325‐330. - PubMed
-
- Quiros‐Tejeira RE, Ament ME, Heyman MB, Martin MG, Rosenthal P, Hall TR, et al. Variable morbidity in alagille syndrome: a review of 43 cases. J Pediatr Gastroenterol Nutr 1999;29:431‐437. - PubMed
-
- Li L, Krantz ID, Deng Y, Genin A, Banta AB, Collins CC, et al. Alagille syndrome is caused by mutations in human Jagged1, which encodes a ligand for Notch1. Nat Genet 1997;16:243‐251. - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
