

Neuroinflammation in the normal-appearing white matter (NAWM) of the multiple sclerosis brain causes abnormalities at the nodes of Ranvier
- PMID: 33315860
- PMCID: PMC7769608
- DOI: 10.1371/journal.pbio.3001008
Neuroinflammation in the normal-appearing white matter (NAWM) of the multiple sclerosis brain causes abnormalities at the nodes of Ranvier
Abstract
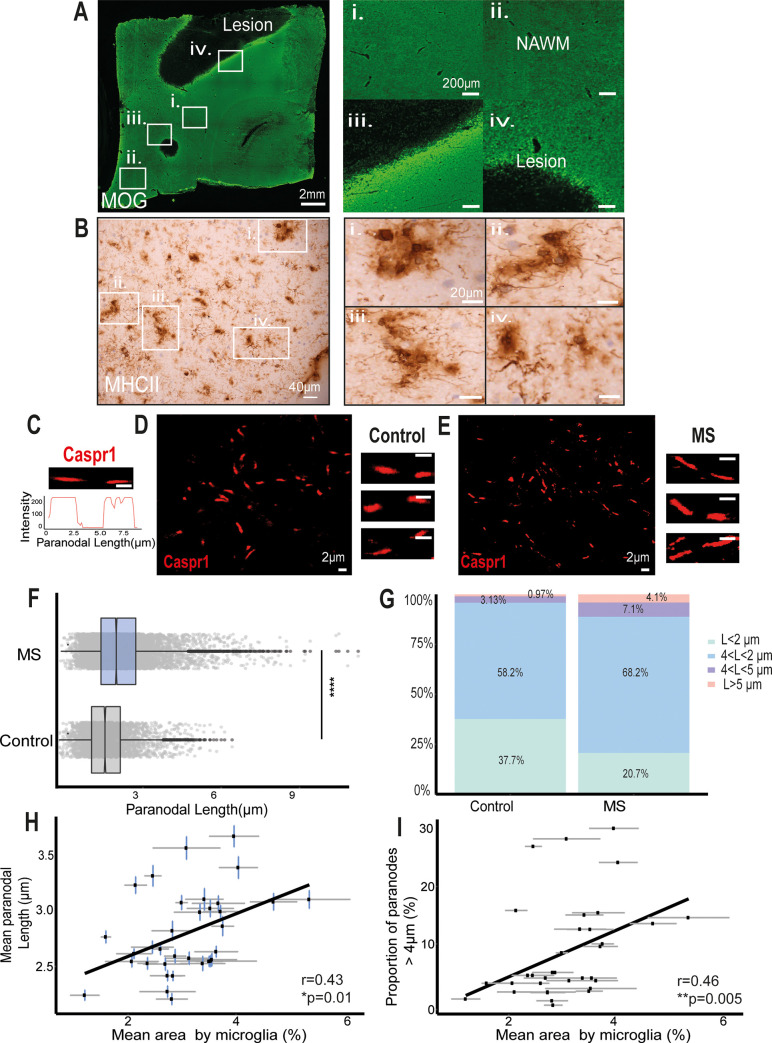
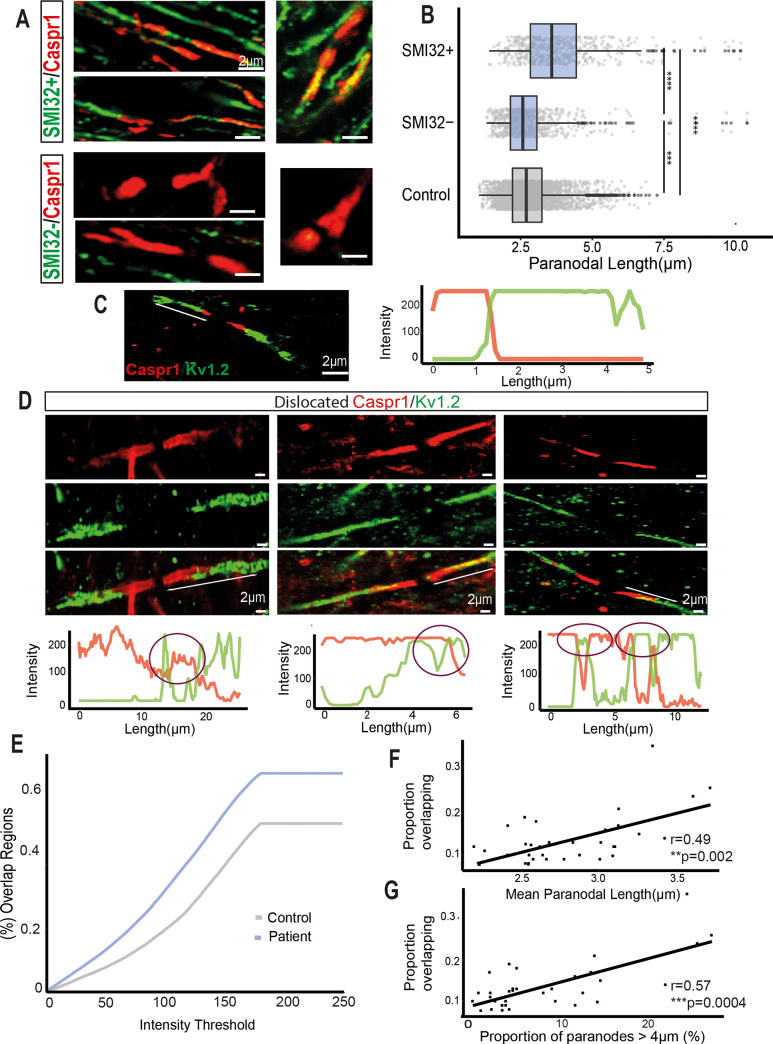
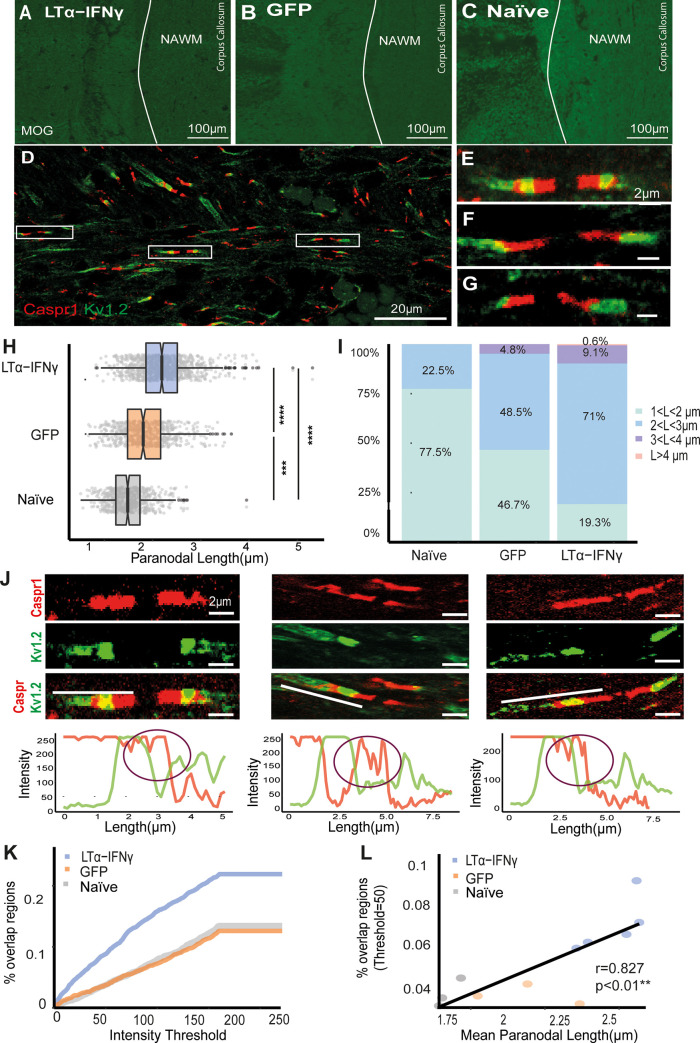
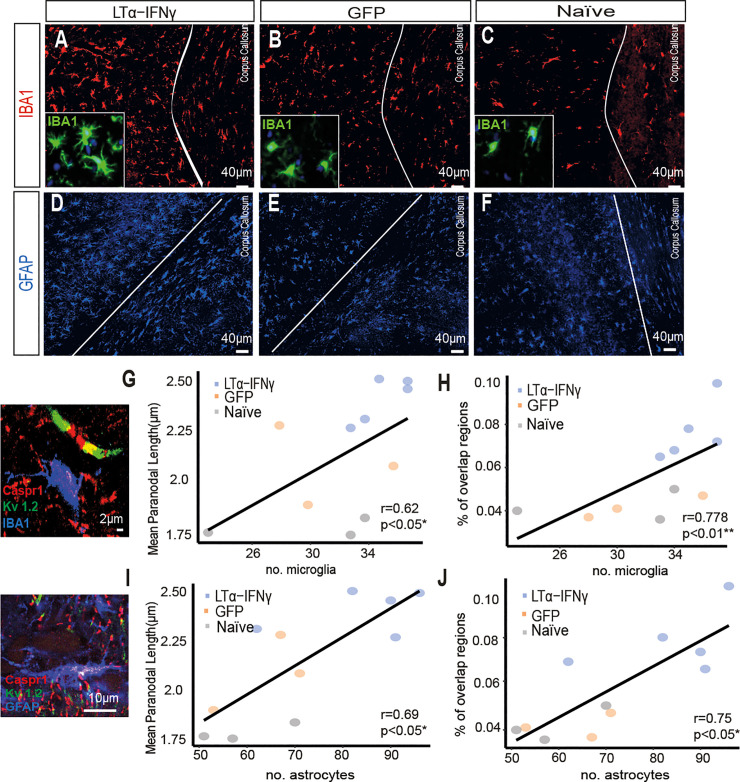
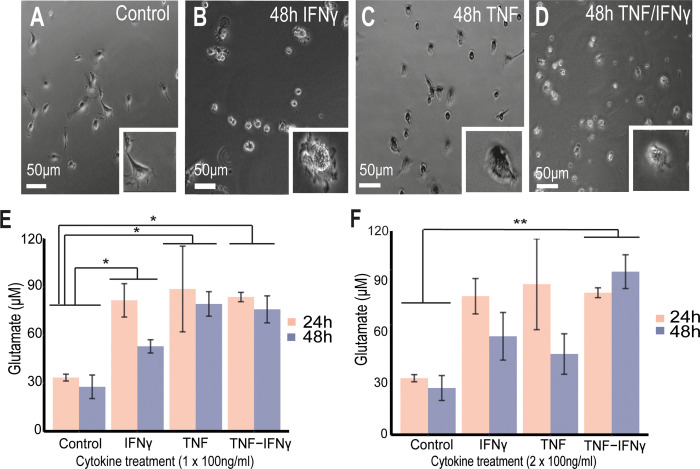
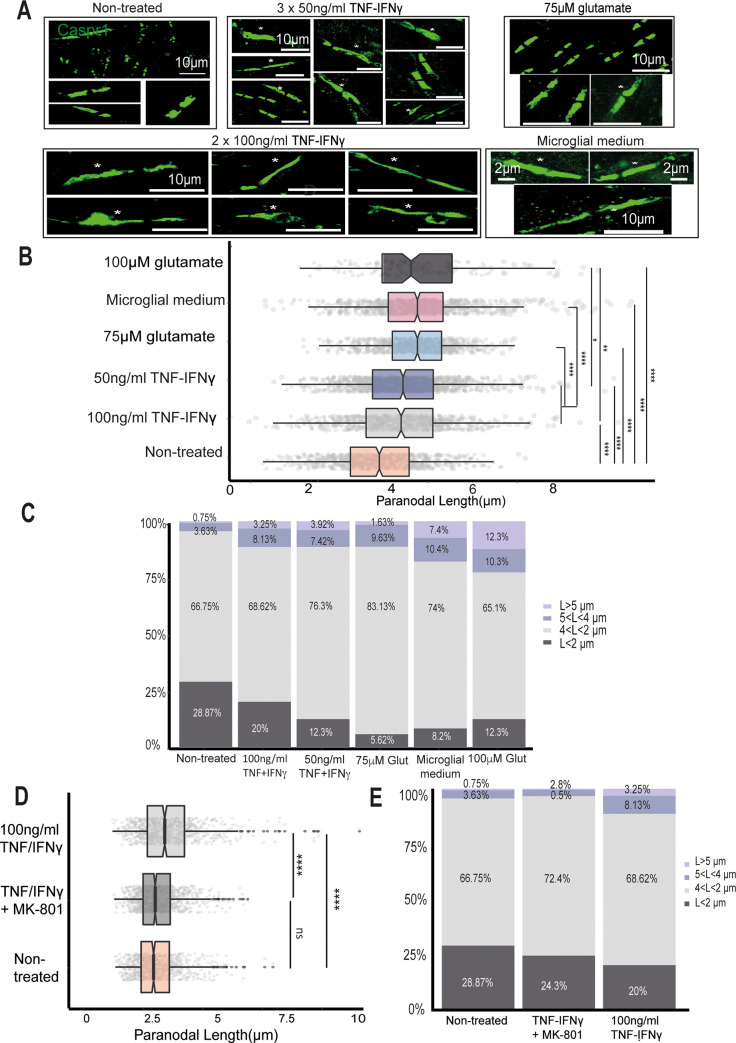
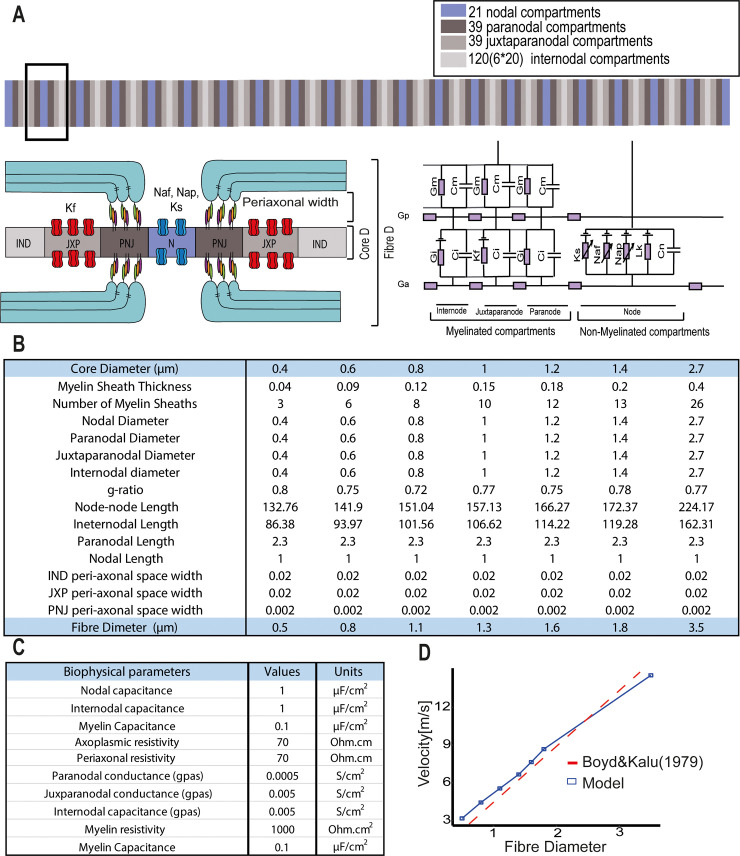
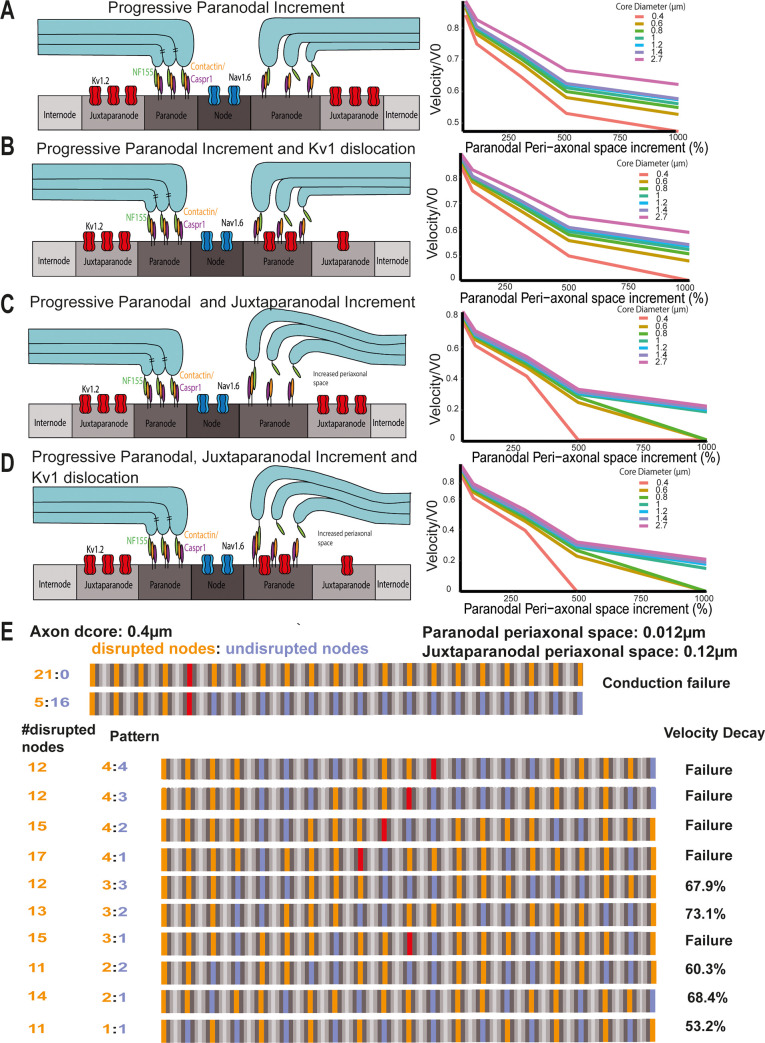
Changes to the structure of nodes of Ranvier in the normal-appearing white matter (NAWM) of multiple sclerosis (MS) brains are associated with chronic inflammation. We show that the paranodal domains in MS NAWM are longer on average than control, with Kv1.2 channels dislocated into the paranode. These pathological features are reproduced in a model of chronic meningeal inflammation generated by the injection of lentiviral vectors for the lymphotoxin-α (LTα) and interferon-γ (IFNγ) genes. We show that tumour necrosis factor (TNF), IFNγ, and glutamate can provoke paranodal elongation in cerebellar slice cultures, which could be reversed by an N-methyl-D-aspartate (NMDA) receptor blocker. When these changes were inserted into a computational model to simulate axonal conduction, a rapid decrease in velocity was observed, reaching conduction failure in small diameter axons. We suggest that glial cells activated by pro-inflammatory cytokines can produce high levels of glutamate, which triggers paranodal pathology, contributing to axonal damage and conduction deficits.
Conflict of interest statement
I have read the journal's policy and the authors of this manuscript have the following competing interests: PG, RJ, EB, JM, SU and CP have no competing interests. RR has received research funds from MedImmune plc and consultancy fees from Roche and Novartis. OWH has received consultancy fees from Roche.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
