

SWI/SNF Complex Mutations Promote Thyroid Tumor Progression and Insensitivity to Redifferentiation Therapies
- PMID: 33318036
- PMCID: PMC8102308
- DOI: 10.1158/2159-8290.CD-20-0735
SWI/SNF Complex Mutations Promote Thyroid Tumor Progression and Insensitivity to Redifferentiation Therapies
Abstract
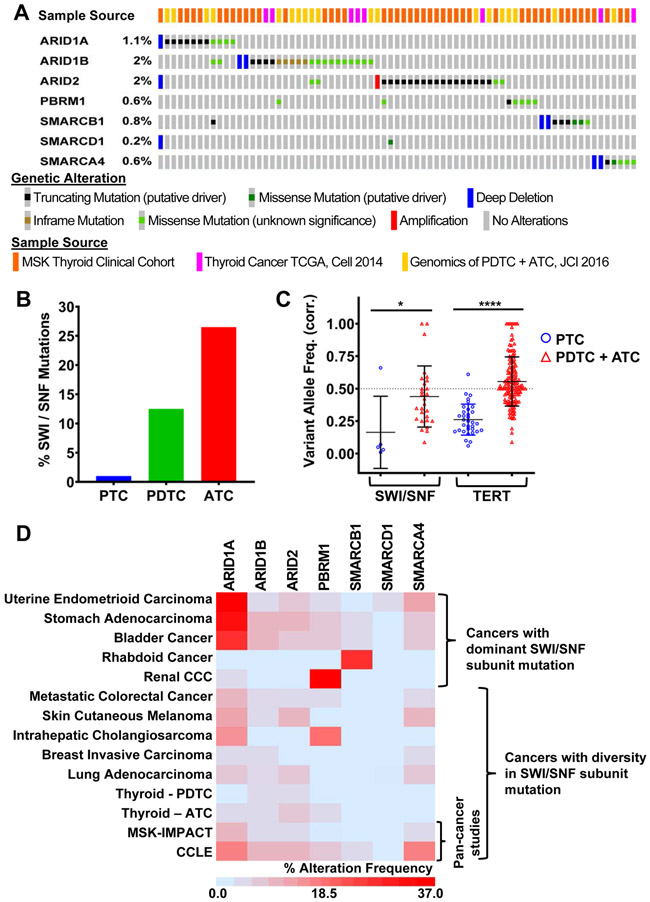
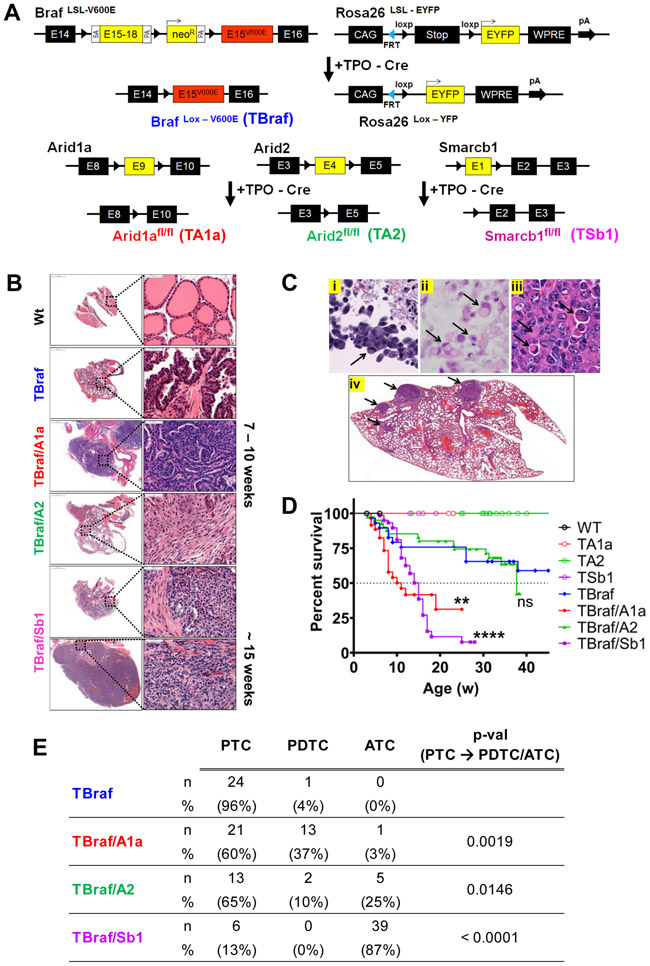
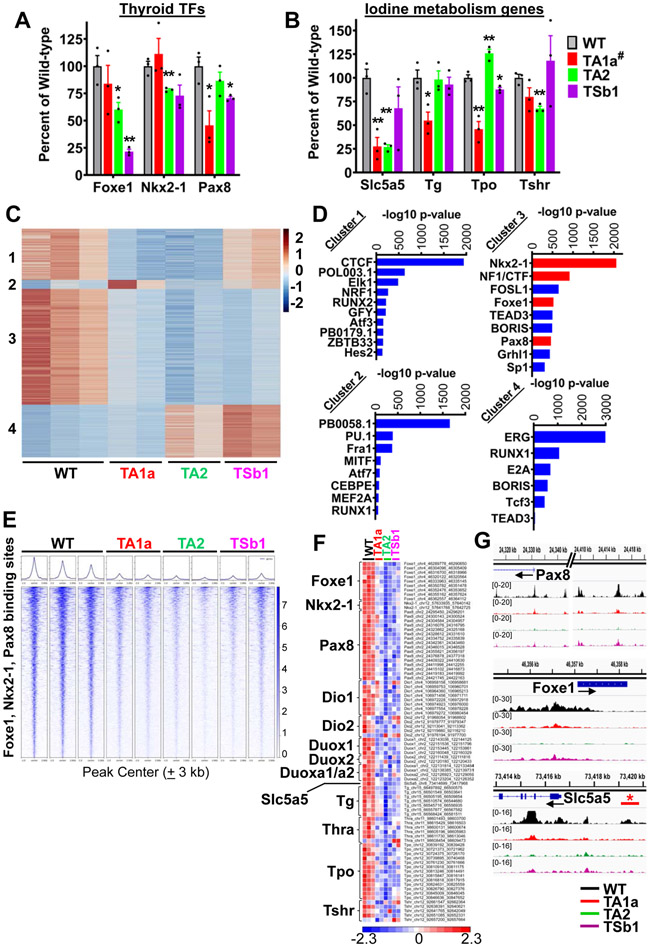
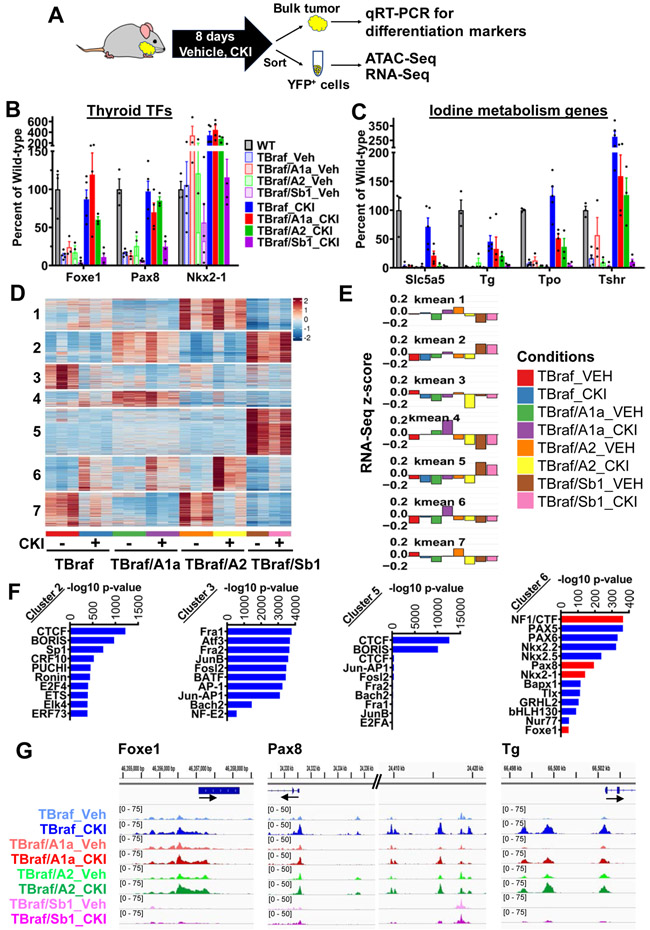
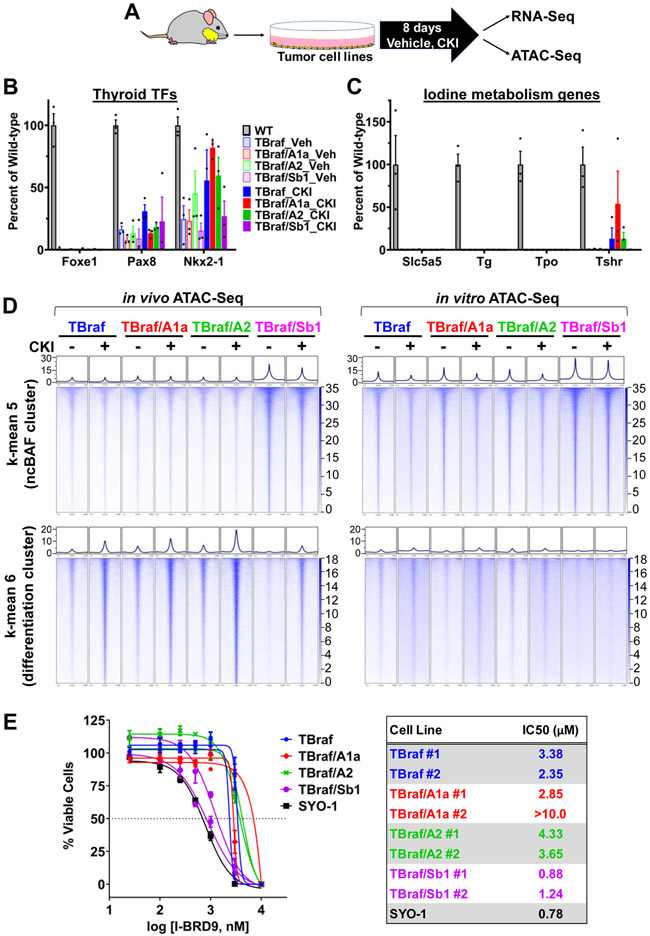
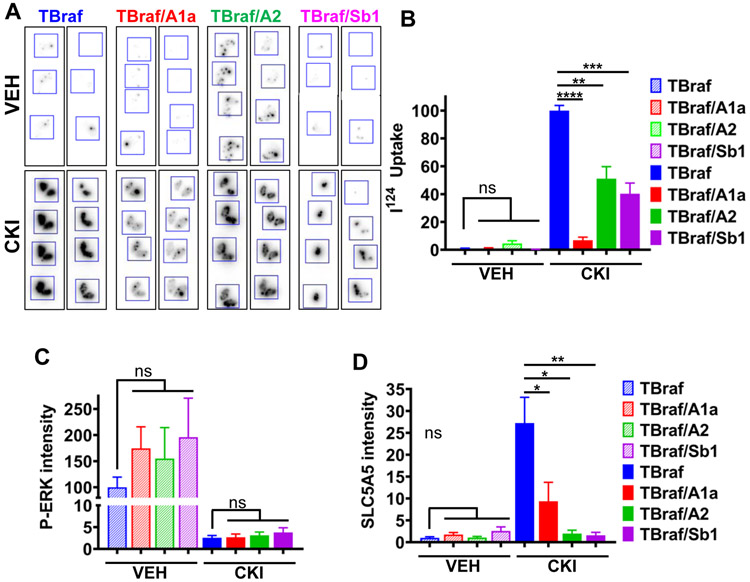
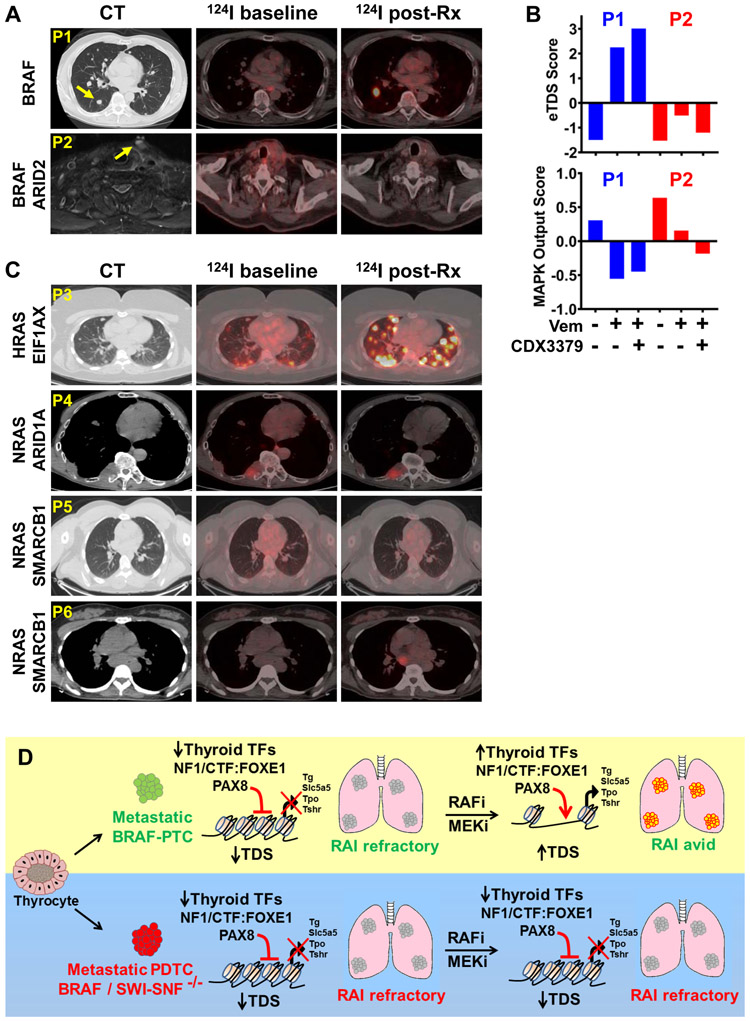
Mutations of subunits of the SWI/SNF chromatin remodeling complexes occur commonly in cancers of different lineages, including advanced thyroid cancers. Here we show that thyroid-specific loss of Arid1a, Arid2, or Smarcb1 in mouse BRAFV600E-mutant tumors promotes disease progression and decreased survival, associated with lesion-specific effects on chromatin accessibility and differentiation. As compared with normal thyrocytes, BRAFV600E-mutant mouse papillary thyroid cancers have decreased lineage transcription factor expression and accessibility to their target DNA binding sites, leading to impairment of thyroid-differentiated gene expression and radioiodine incorporation, which is rescued by MAPK inhibition. Loss of individual SWI/SNF subunits in BRAF tumors leads to a repressive chromatin state that cannot be reversed by MAPK pathway blockade, rendering them insensitive to its redifferentiation effects. Our results show that SWI/SNF complexes are central to the maintenance of differentiated function in thyroid cancers, and their loss confers radioiodine refractoriness and resistance to MAPK inhibitor-based redifferentiation therapies. SIGNIFICANCE: Reprogramming cancer differentiation confers therapeutic benefit in various disease contexts. Oncogenic BRAF silences genes required for radioiodine responsiveness in thyroid cancer. Mutations in SWI/SNF genes result in loss of chromatin accessibility at thyroid lineage specification genes in BRAF-mutant thyroid tumors, rendering them insensitive to the redifferentiation effects of MAPK blockade.This article is highlighted in the In This Issue feature, p. 995.
©2020 American Association for Cancer Research.
Conflict of interest statement
Conflict of interest
JAF: Consultant, Loxo Oncology. Grant support, Eisai Pharmaceuticals.
ALH: Research Funding (Clinical trials): AstraZeneca, Novartis, Eisai Pharmaceuticals, Genentech-Roche, Bayer, Celldex (previously Koltan). Advisory Board: AstraZeneca, Eisai Pharmaceuticals, Sanofi Genzyme, Novartis, Genentech-Roche.
Figures
References
-
- Ward PS, Patel J, Wise DR, Abdel-Wahab O, Bennett BD, Coller HA, et al. The Common Feature of Leukemia-Associated IDH1 and IDH2 Mutations Is a Neomorphic Enzyme Activity Converting alpha-Ketoglutarate to 2-Hydroxyglutarate. Cancer Cell 2010;17(3):225–34 doi 10.1016/j.ccr.2010.01.020. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
