

Durable Suppression of Acquired MEK Inhibitor Resistance in Cancer by Sequestering MEK from ERK and Promoting Antitumor T-cell Immunity
- PMID: 33318037
- PMCID: PMC7933113
- DOI: 10.1158/2159-8290.CD-20-0873
Durable Suppression of Acquired MEK Inhibitor Resistance in Cancer by Sequestering MEK from ERK and Promoting Antitumor T-cell Immunity
Expression of concern in
-
Expression of Concern: Durable Suppression of Acquired MEK Inhibitor Resistance in Cancer by Sequestering MEK from ERK and Promoting Antitumor T-cell Immunity.Cancer Discov. 2024 Jul 1;14(7):1357. doi: 10.1158/2159-8290.CD-24-0613. Cancer Discov. 2024. PMID: 38946326 No abstract available.
Abstract
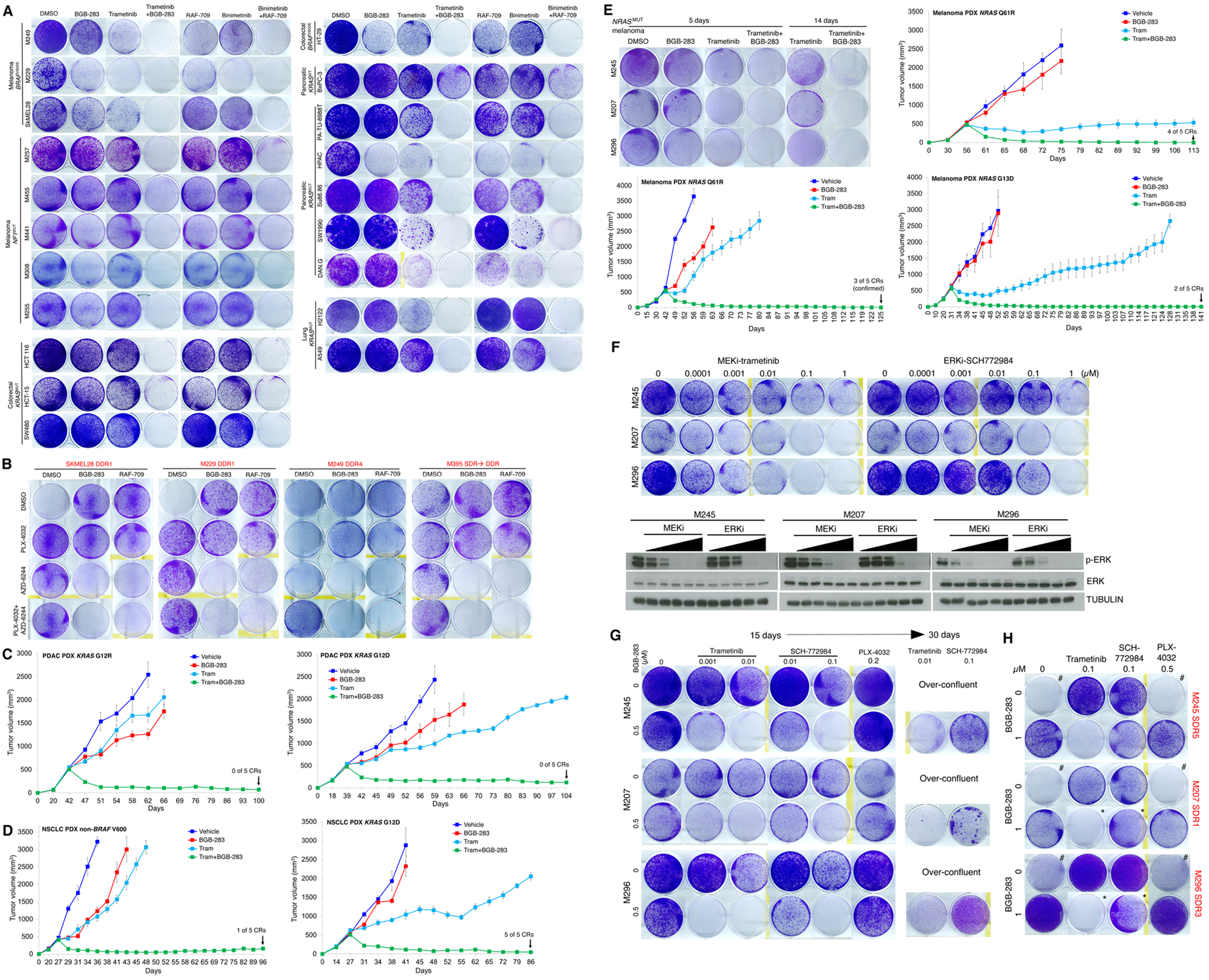
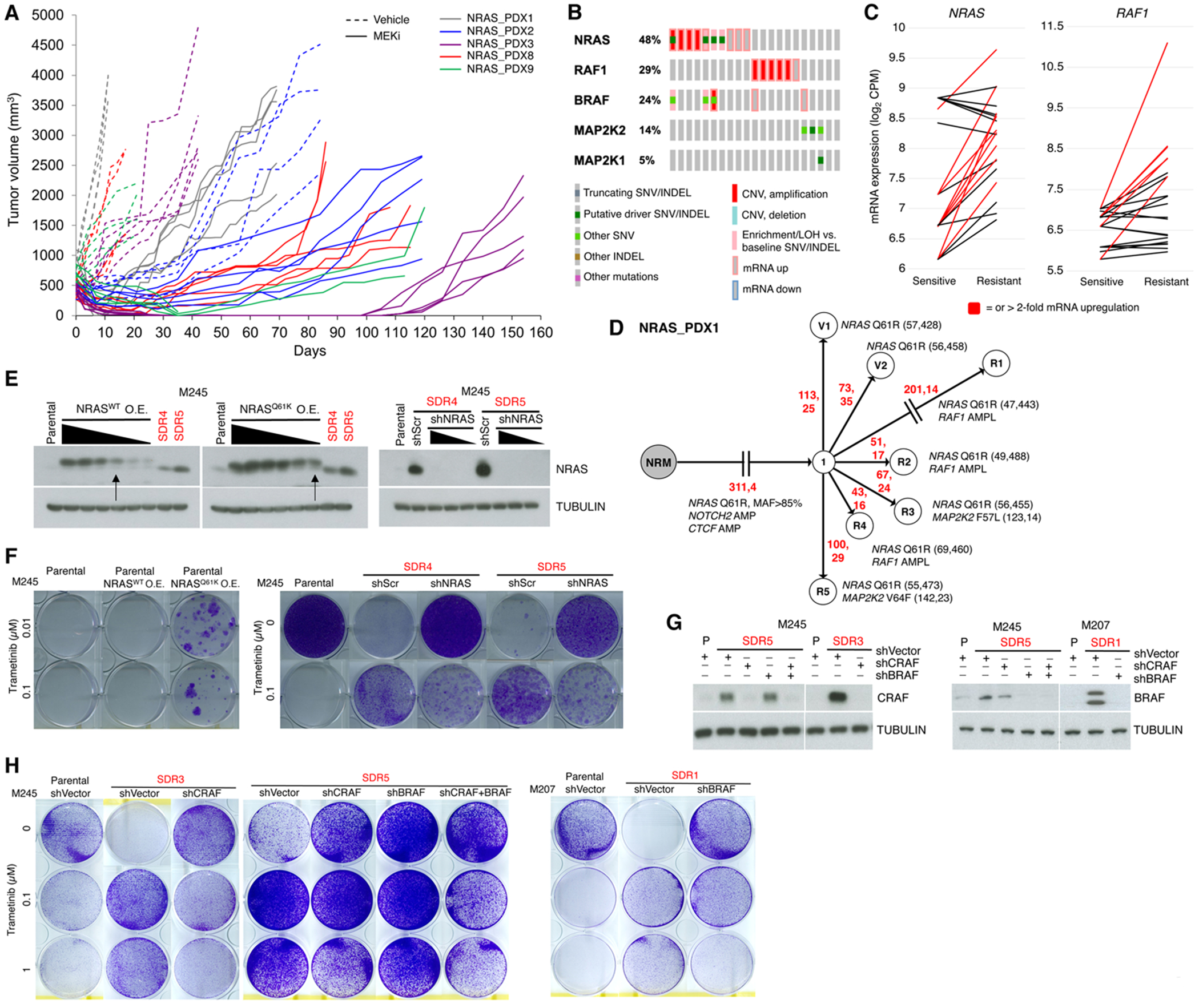
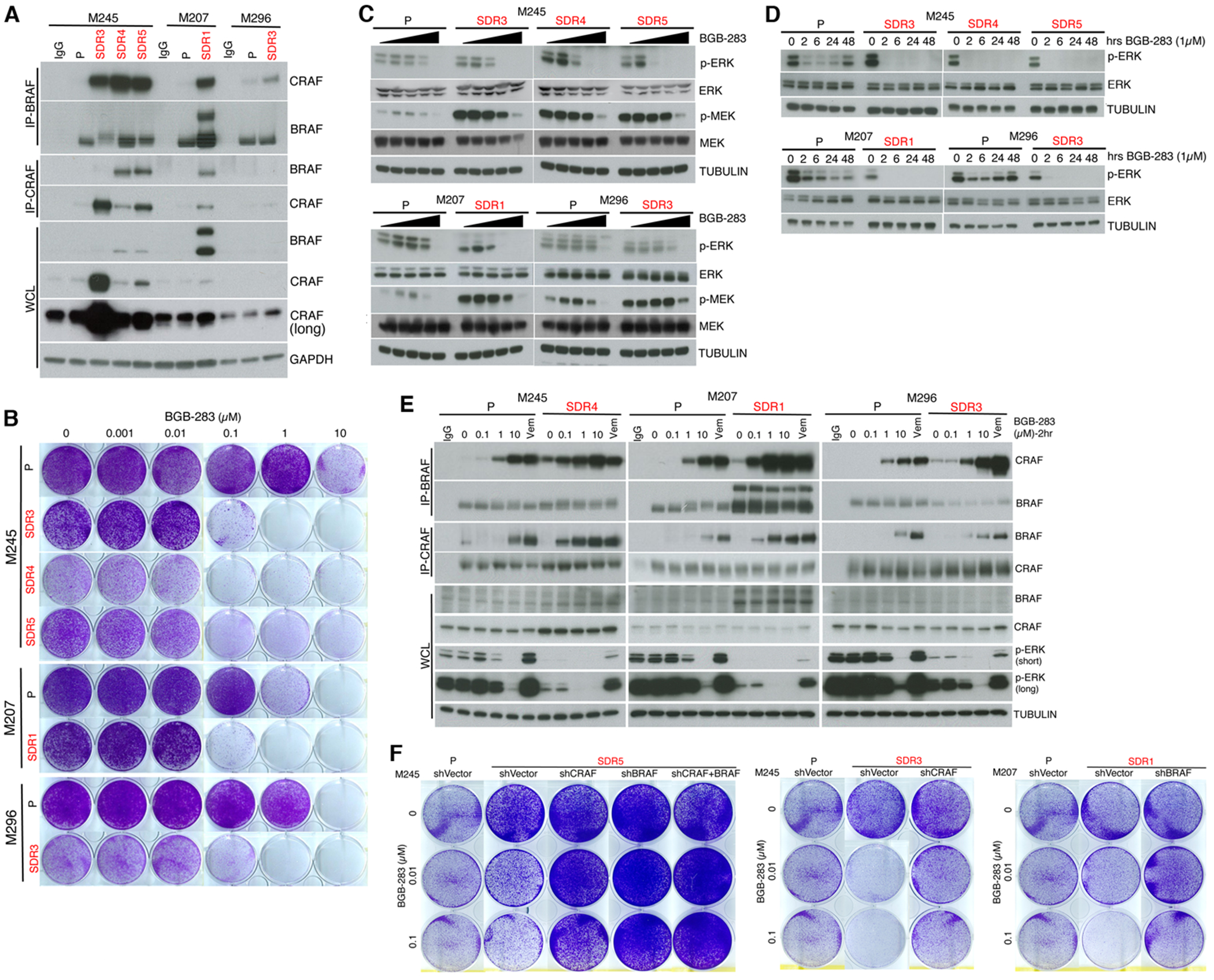
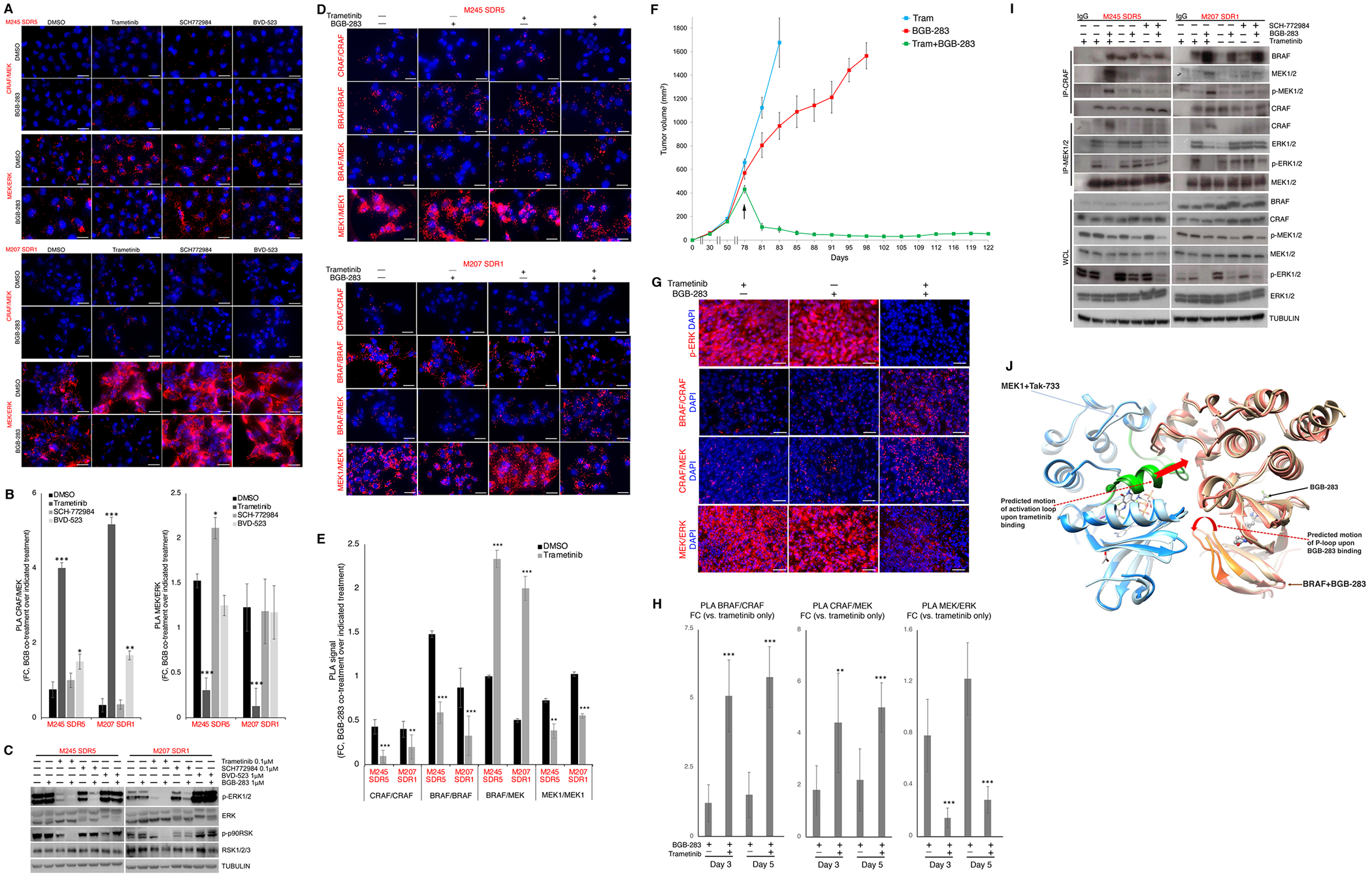
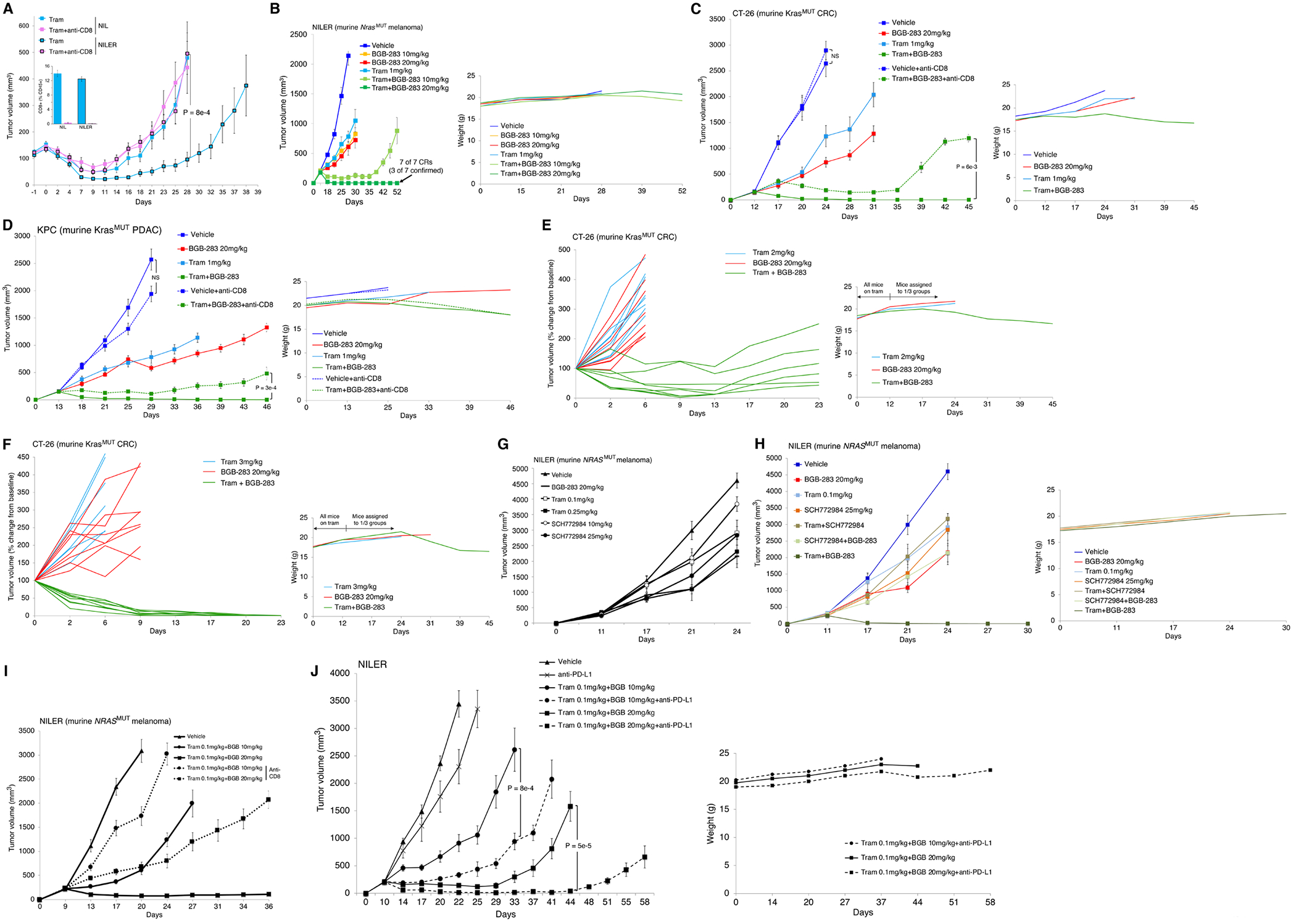
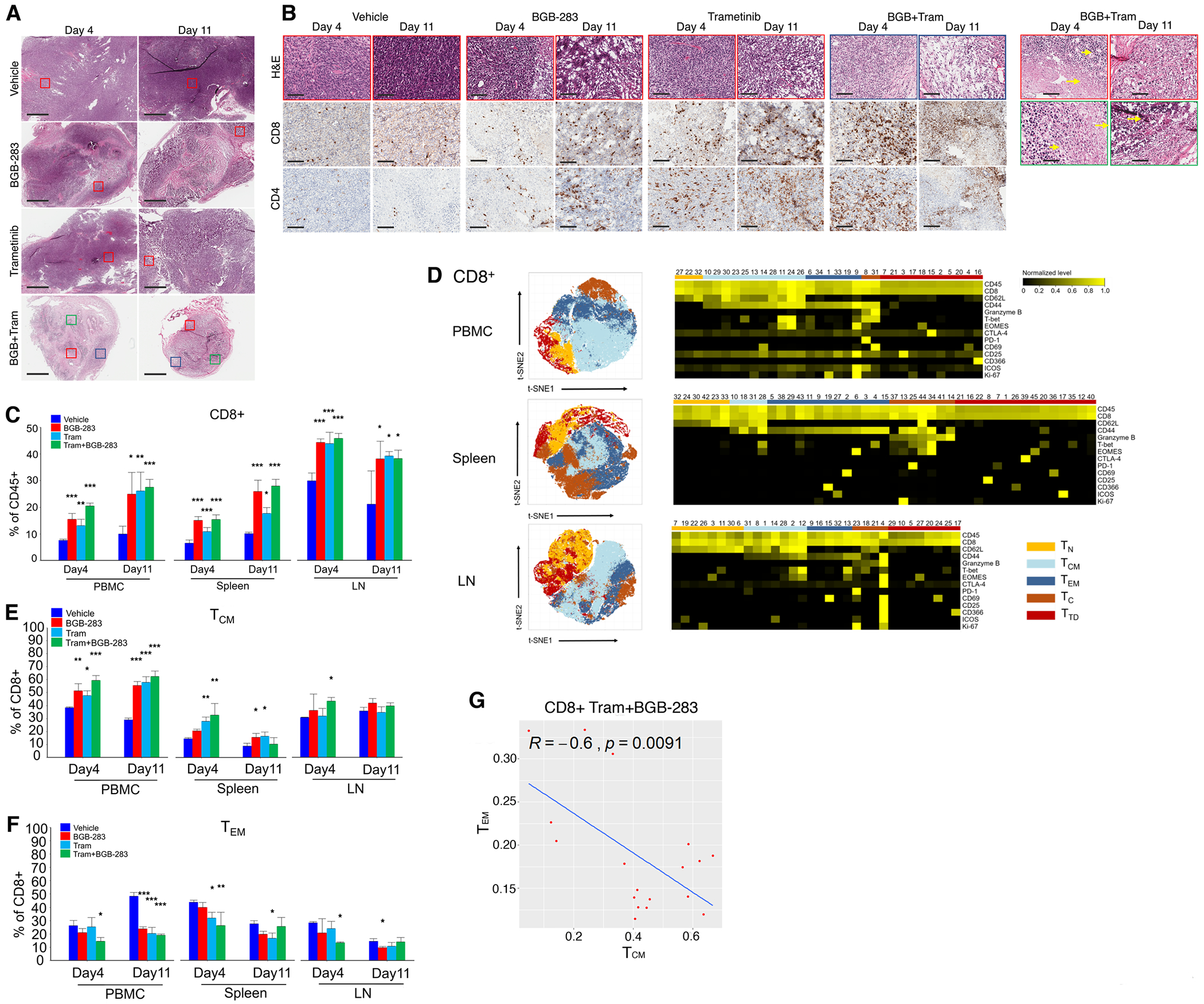
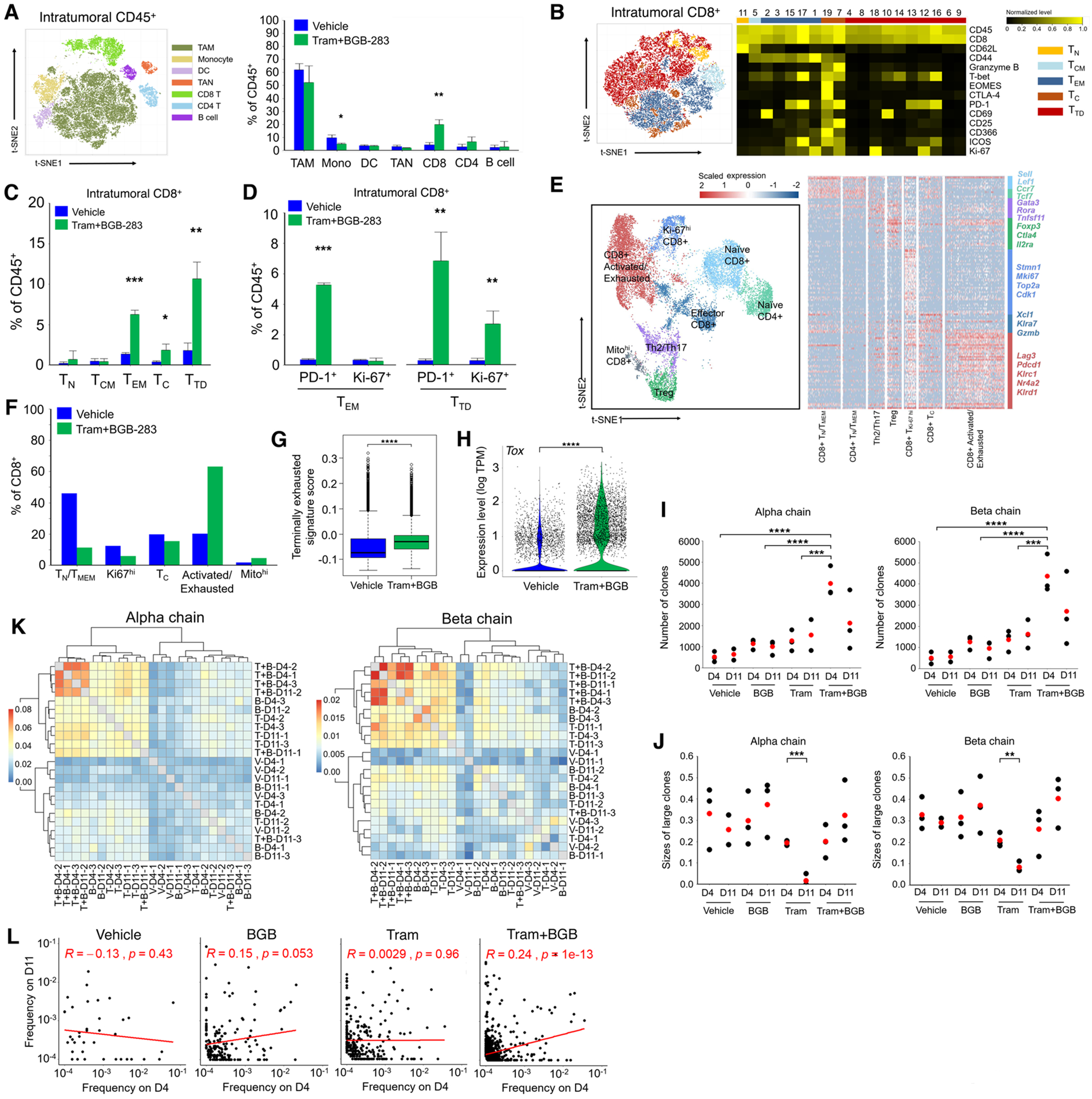
MAPK targeting in cancer often fails due to MAPK reactivation. MEK inhibitor (MEKi) monotherapy provides limited clinical benefits but may serve as a foundation for combination therapies. Here, we showed that combining a type II RAF inhibitor (RAFi) with an allosteric MEKi durably prevents and overcomes acquired resistance among cancers with KRAS, NRAS, NF1, BRAF non-V600, and BRAF V600 mutations. Tumor cell-intrinsically, type II RAFi plus MEKi sequester MEK in RAF complexes, reduce MEK/MEK dimerization, and uncouple MEK from ERK in acquired-resistant tumor subpopulations. Immunologically, this combination expands memory and activated/exhausted CD8+ T cells, and durable tumor regression elicited by this combination requires CD8+ T cells, which can be reinvigorated by anti-PD-L1 therapy. Whereas MEKi reduces dominant intratumoral T-cell clones, type II RAFi cotreatment reverses this effect and promotes T-cell clonotypic expansion. These findings rationalize the clinical development of type II RAFi plus MEKi and their further combination with PD-1/L1-targeted therapy. SIGNIFICANCE: Type I RAFi + MEKi are indicated only in certain BRAF V600MUT cancers. In contrast, type II RAFi + MEKi are durably active against acquired MEKi resistance across broad cancer indications, which reveals exquisite MAPK addiction. Allosteric modulation of MAPK protein/protein interactions and temporal preservation of intratumoral CD8+ T cells are mechanisms that may be further exploited.This article is highlighted in the In This Issue feature, p. 521.
©2020 American Association for Cancer Research.
Figures
References
-
- Samatar AA, Poulikakos PI. Targeting RAS-ERK signalling in cancer: promises and challenges. Nat Rev Drug Discov 2014;13:928–42 - PubMed
-
- Dummer R, Schadendorf D, Ascierto PA, Arance A, Dutriaux C, Di Giacomo AM, et al. Binimetinib versus dacarbazine in patients with advanced NRAS-mutant melanoma (NEMO): a multicentre, open-label, randomised, phase 3 trial. Lancet Oncol 2017;18:435–45 - PubMed
-
- Flaherty KT, Robert C, Hersey P, Nathan P, Garbe C, Milhem M, et al. Improved survival with MEK inhibition in BRAF-mutated melanoma. N Engl J Med 2012;367:107–14 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
