

Efficient hybrid de novo assembly of human genomes with WENGAN
- PMID: 33318652
- PMCID: PMC8041623
- DOI: 10.1038/s41587-020-00747-w
Efficient hybrid de novo assembly of human genomes with WENGAN
Abstract
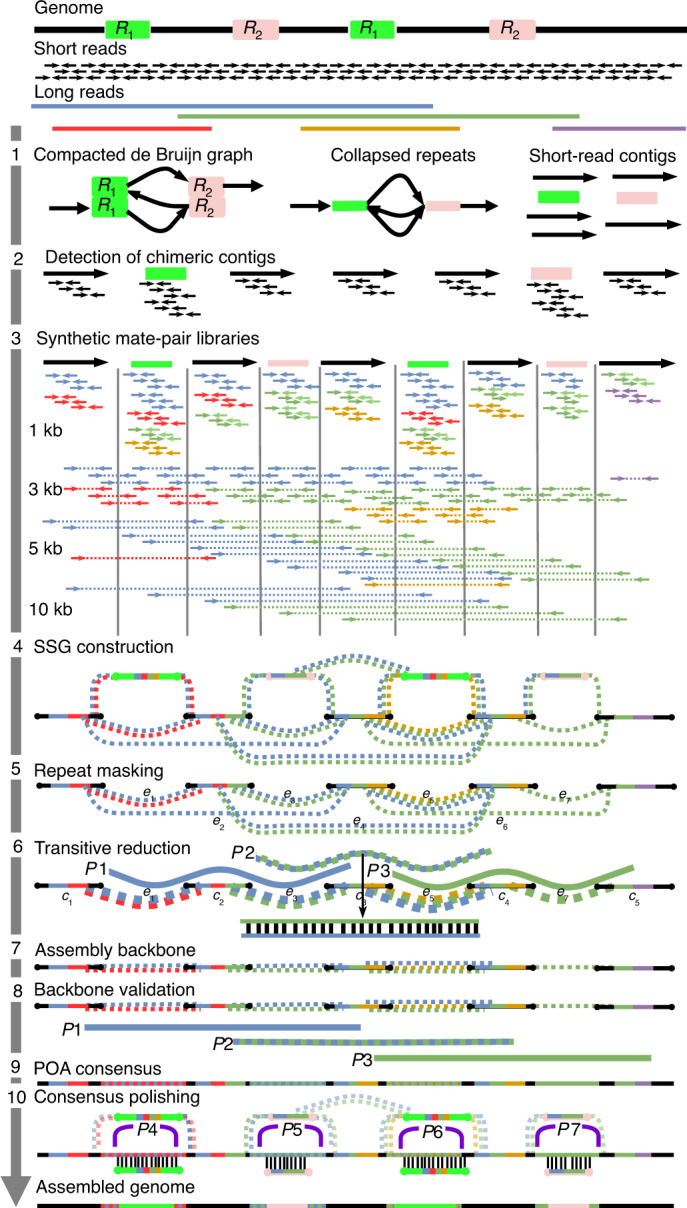
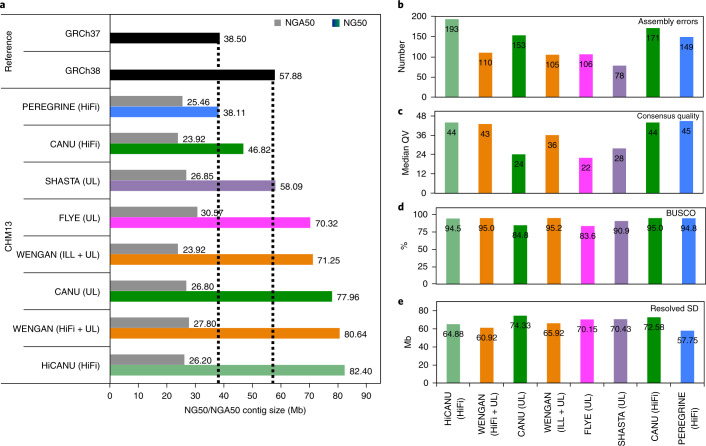
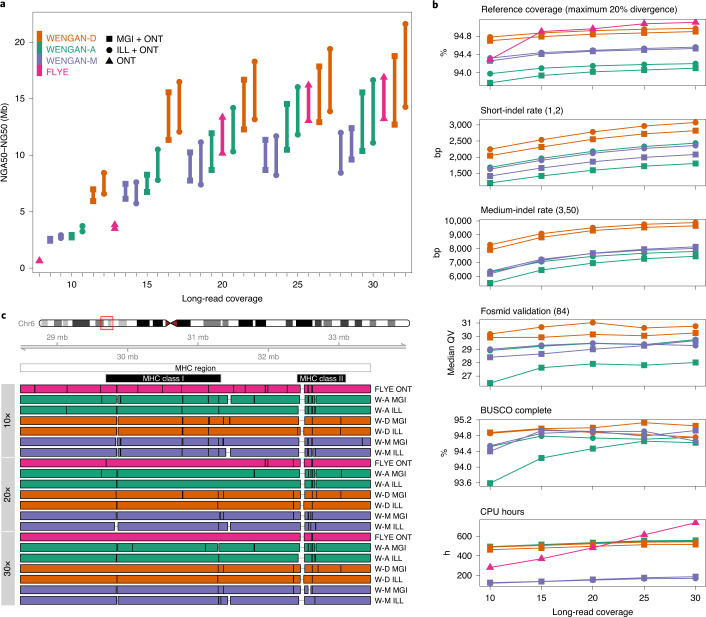
Generating accurate genome assemblies of large, repeat-rich human genomes has proved difficult using only long, error-prone reads, and most human genomes assembled from long reads add accurate short reads to polish the consensus sequence. Here we report an algorithm for hybrid assembly, WENGAN, that provides very high quality at low computational cost. We demonstrate de novo assembly of four human genomes using a combination of sequencing data generated on ONT PromethION, PacBio Sequel, Illumina and MGI technology. WENGAN implements efficient algorithms to improve assembly contiguity as well as consensus quality. The resulting genome assemblies have high contiguity (contig NG50: 17.24-80.64 Mb), few assembly errors (contig NGA50: 11.8-59.59 Mb), good consensus quality (QV: 27.84-42.88) and high gene completeness (BUSCO complete: 94.6-95.2%), while consuming low computational resources (CPU hours: 187-1,200). In particular, the WENGAN assembly of the haploid CHM13 sample achieved a contig NG50 of 80.64 Mb (NGA50: 59.59 Mb), which surpasses the contiguity of the current human reference genome (GRCh38 contig NG50: 57.88 Mb).
Conflict of interest statement
The authors declare no competing interests.
Figures

References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
