

A collection of bacterial isolates from the pig intestine reveals functional and taxonomic diversity
- PMID: 33319778
- PMCID: PMC7738495
- DOI: 10.1038/s41467-020-19929-w
A collection of bacterial isolates from the pig intestine reveals functional and taxonomic diversity
Abstract
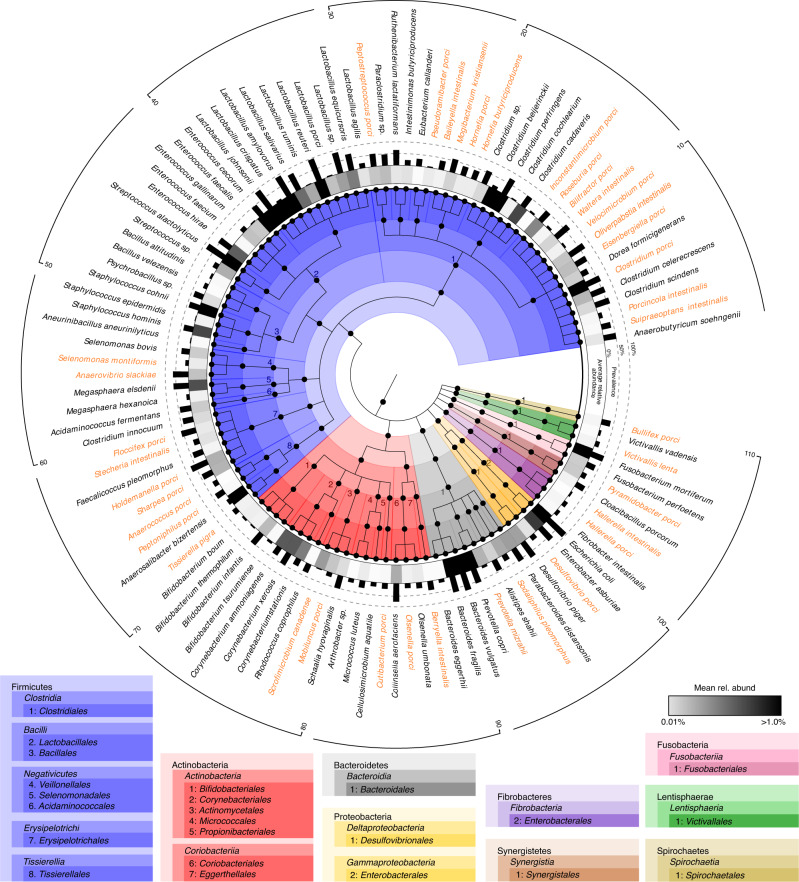
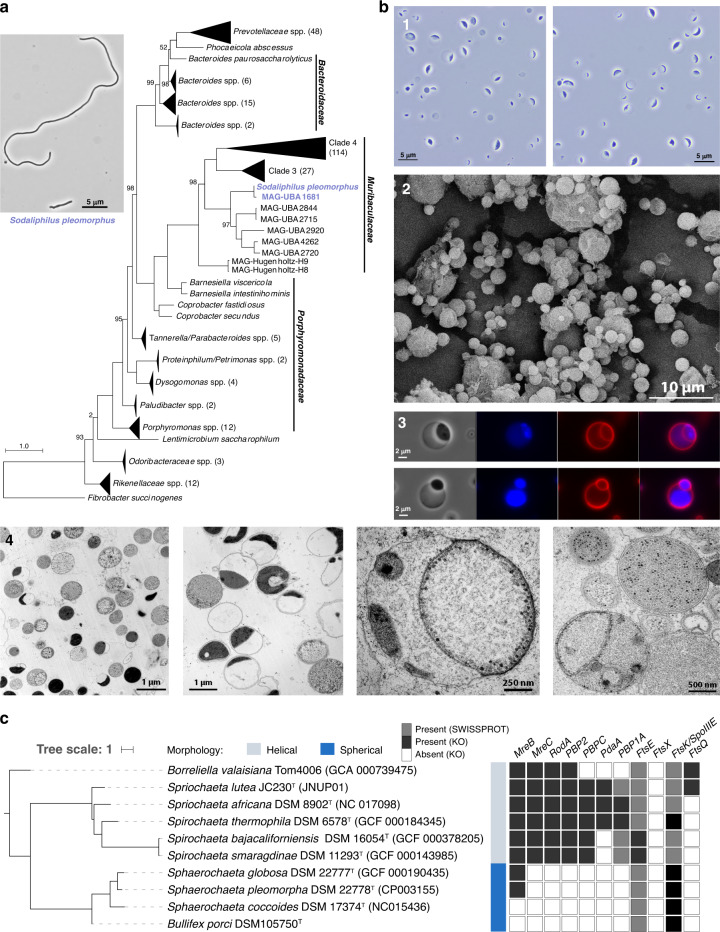
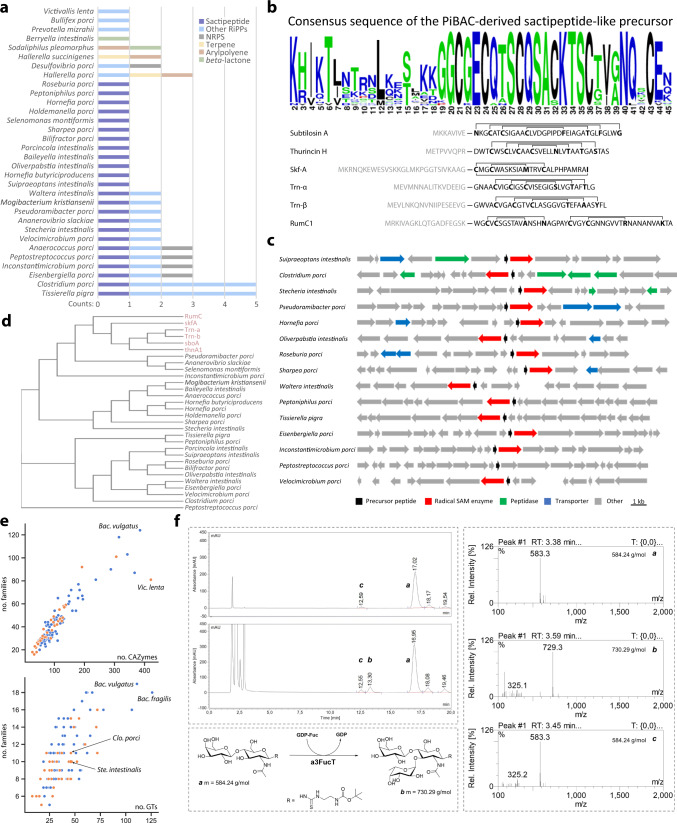
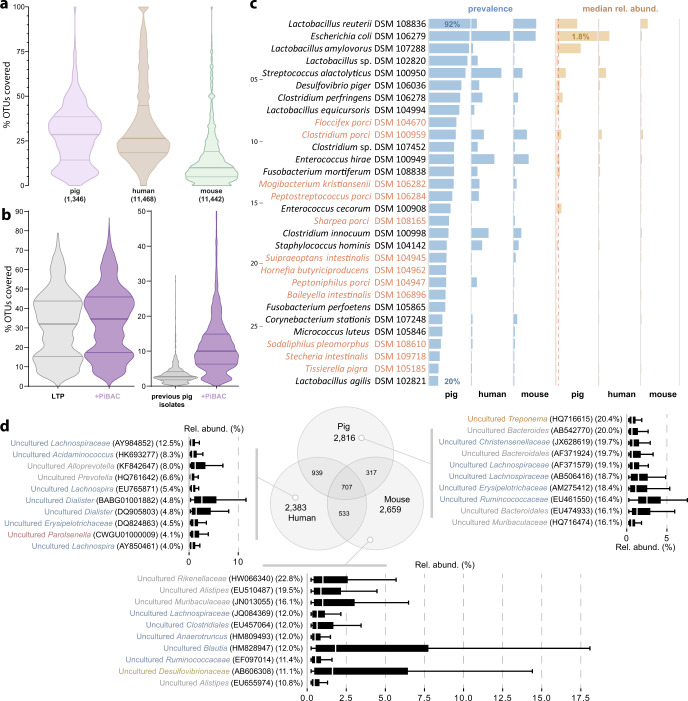
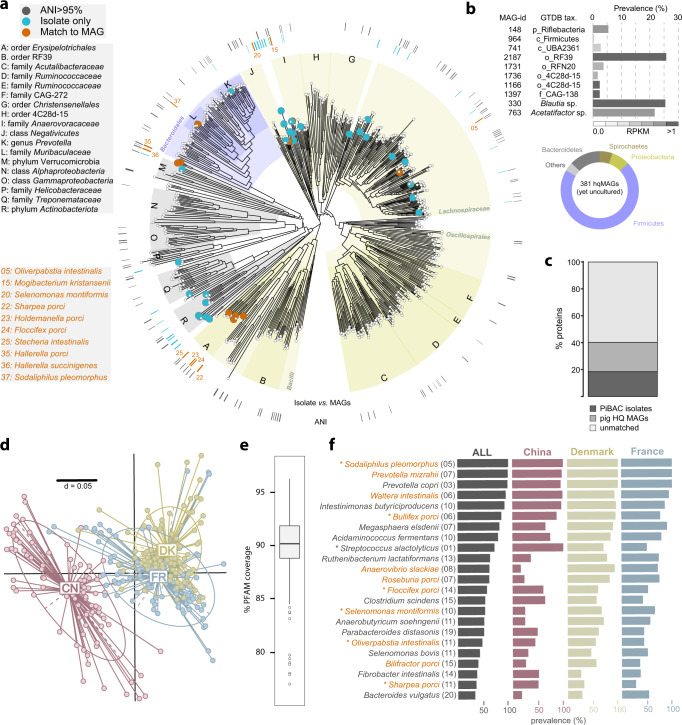
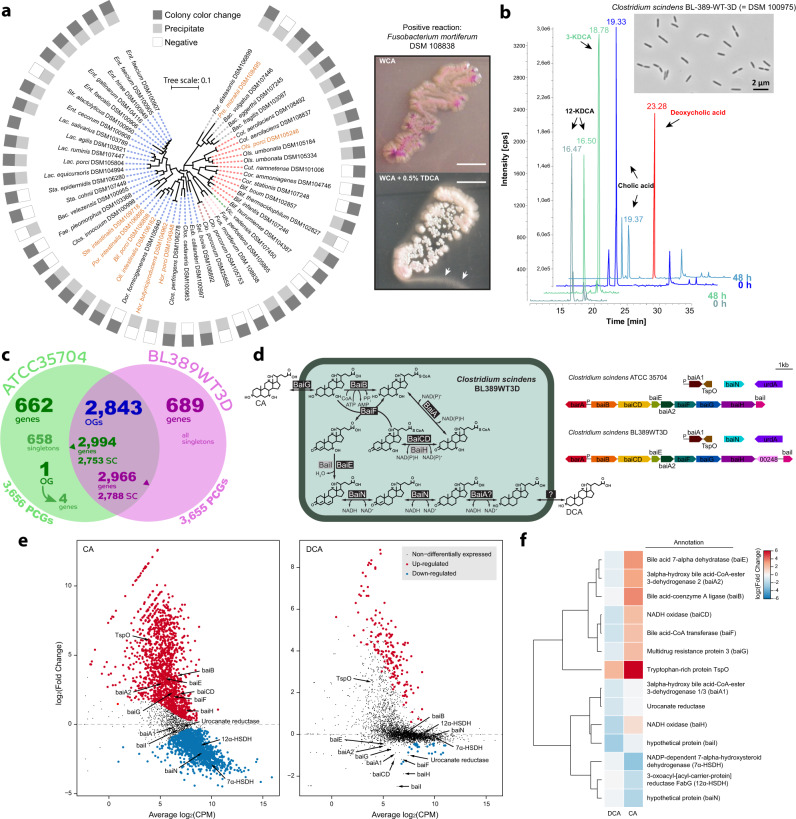
Our knowledge about the gut microbiota of pigs is still scarce, despite the importance of these animals for biomedical research and agriculture. Here, we present a collection of cultured bacteria from the pig gut, including 110 species across 40 families and nine phyla. We provide taxonomic descriptions for 22 novel species and 16 genera. Meta-analysis of 16S rRNA amplicon sequence data and metagenome-assembled genomes reveal prevalent and pig-specific species within Lactobacillus, Streptococcus, Clostridium, Desulfovibrio, Enterococcus, Fusobacterium, and several new genera described in this study. Potentially interesting functions discovered in these organisms include a fucosyltransferase encoded in the genome of the novel species Clostridium porci, and prevalent gene clusters for biosynthesis of sactipeptide-like peptides. Many strains deconjugate primary bile acids in in vitro assays, and a Clostridium scindens strain produces secondary bile acids via dehydroxylation. In addition, cells of the novel species Bullifex porci are coccoidal or spherical under the culture conditions tested, in contrast with the usual helical shape of other members of the family Spirochaetaceae. The strain collection, called 'Pig intestinal bacterial collection' (PiBAC), is publicly available at www.dsmz.de/pibac and opens new avenues for functional studies of the pig gut microbiota.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
