

Comparison of the two up-to-date sequencing technologies for genome assembly: HiFi reads of Pacific Biosciences Sequel II system and ultralong reads of Oxford Nanopore
- PMID: 33319909
- PMCID: PMC7736813
- DOI: 10.1093/gigascience/giaa123
Comparison of the two up-to-date sequencing technologies for genome assembly: HiFi reads of Pacific Biosciences Sequel II system and ultralong reads of Oxford Nanopore
Abstract
Background: The availability of reference genomes has revolutionized the study of biology. Multiple competing technologies have been developed to improve the quality and robustness of genome assemblies during the past decade. The 2 widely used long-read sequencing providers-Pacific Biosciences (PacBio) and Oxford Nanopore Technologies (ONT)-have recently updated their platforms: PacBio enables high-throughput HiFi reads with base-level resolution of >99%, and ONT generated reads as long as 2 Mb. We applied the 2 up-to-date platforms to a single rice individual and then compared the 2 assemblies to investigate the advantages and limitations of each.
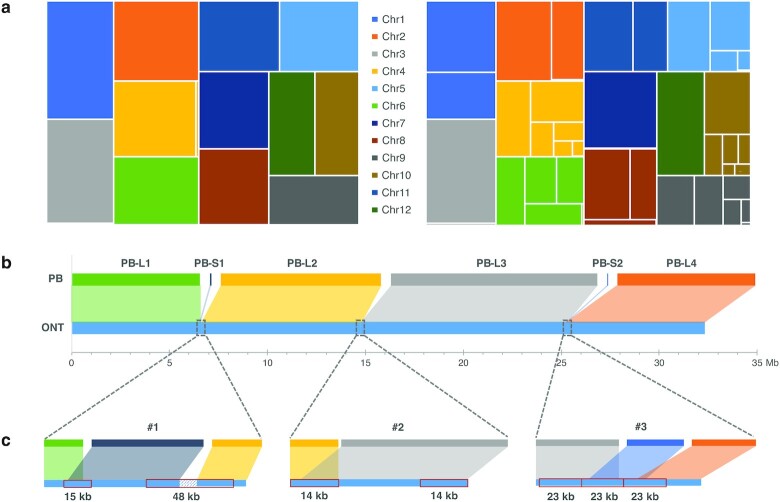
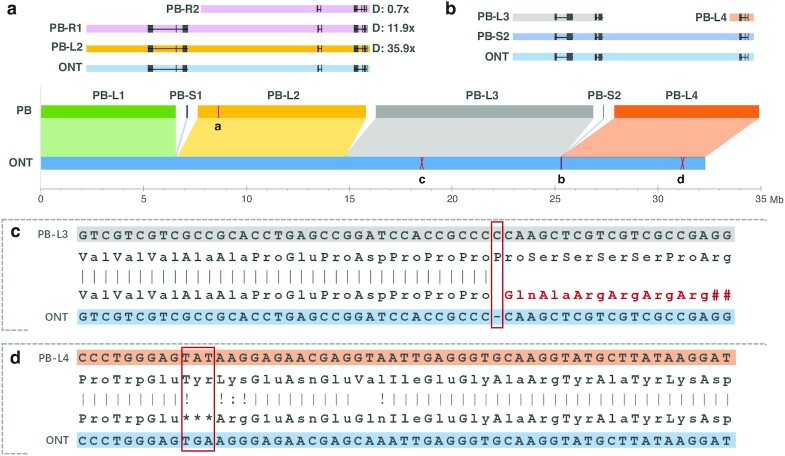
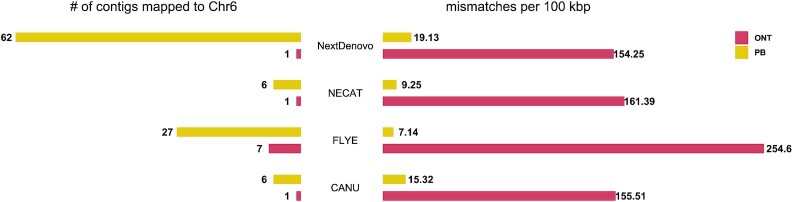
Results: The results showed that ONT ultralong reads delivered higher contiguity, producing a total of 18 contigs of which 10 were assembled into a single chromosome compared to 394 contigs and 3 chromosome-level contigs for the PacBio assembly. The ONT ultralong reads also prevented assembly errors caused by long repetitive regions, for which we observed a total of 44 genes of false redundancies and 10 genes of false losses in the PacBio assembly, leading to over- or underestimation of the gene families in those long repetitive regions. We also noted that the PacBio HiFi reads generated assemblies with considerably fewer errors at the level of single nucleotides and small insertions and deletions than those of the ONT assembly, which generated an average 1.06 errors per kb and finally engendered 1,475 incorrect gene annotations via altered or truncated protein predictions.
Conclusions: It shows that both PacBio HiFi reads and ONT ultralong reads had their own merits. Further genome reference constructions could leverage both techniques to lessen the impact of assembly errors and subsequent annotation mistakes rooted in each.
Keywords: CCS; ONT ultralong; PacBio HiFi; assembly comparison; contiguity; single-molecular sequencer.
© The Author(s) 2020. Published by Oxford University Press GigaScience.
Conflict of interest statement
D.L., P.R., F.L., Z.S,, G.M., Y.T., X.L., Q.L, L.H., D.W. and S.L. are employees of Grandomics Biosciences, a company that provides bioinformatics and genomics services.
Figures
References
-
- Weischenfeldt J, Symmons O, Spitz F, et al. Phenotypic impact of genomic structural variation: insights from and for human disease. Nat Rev Genet. 2013;14:125–38. - PubMed
-
- Fujimoto A, Furuta M, Totoki Y, et al. Whole-genome mutational landscape and characterization of noncoding and structural mutations in liver cancer. Nat Genet. 2016;48:500. - PubMed
-
- Chen YH, Gols R, Benrey B. Crop domestication and its impact on naturally selected trophic interactions. Annu Rev Entomol. 2015;60:35–58. - PubMed
-
- Wheeler DA, Srinivasan M, Egholm M, et al. The complete genome of an individual by massively parallel DNA sequencing. Nature. 2008;452:872–6. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Miscellaneous
