

Bayesian Inference Associates Rare KDR Variants with Specific Phenotypes in Pulmonary Arterial Hypertension
- PMID: 33320693
- PMCID: PMC7892262
- DOI: 10.1161/CIRCGEN.120.003155
Bayesian Inference Associates Rare KDR Variants with Specific Phenotypes in Pulmonary Arterial Hypertension
Abstract
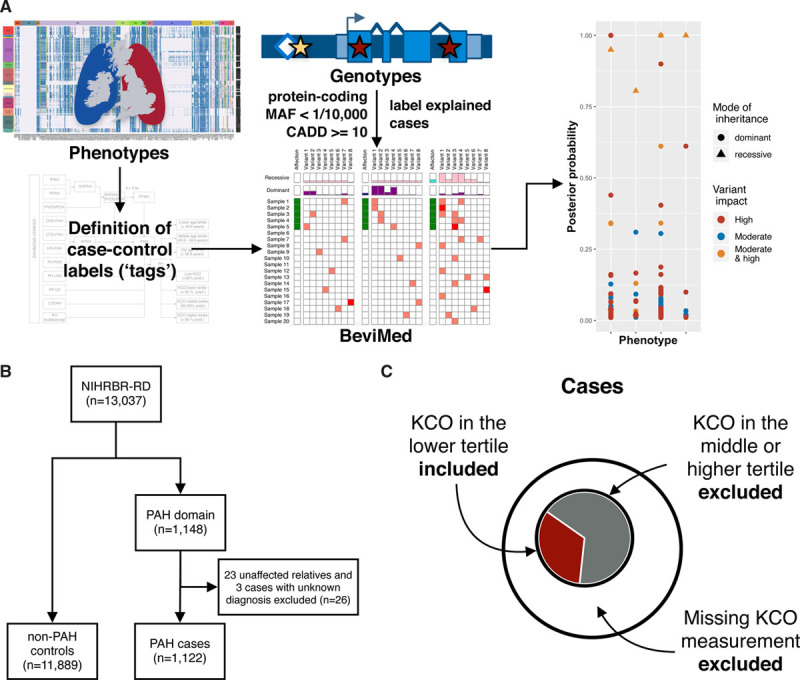
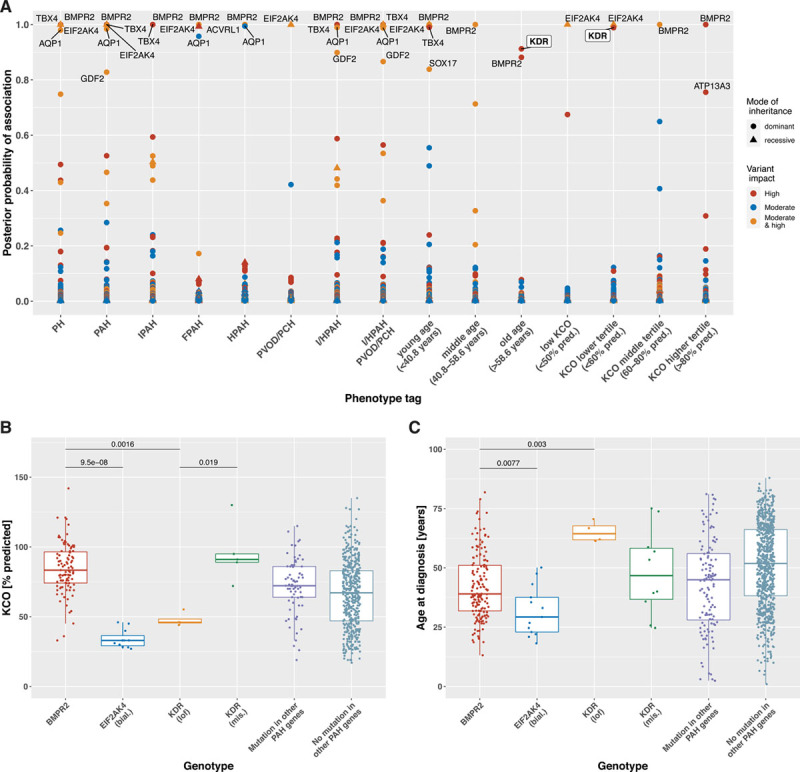
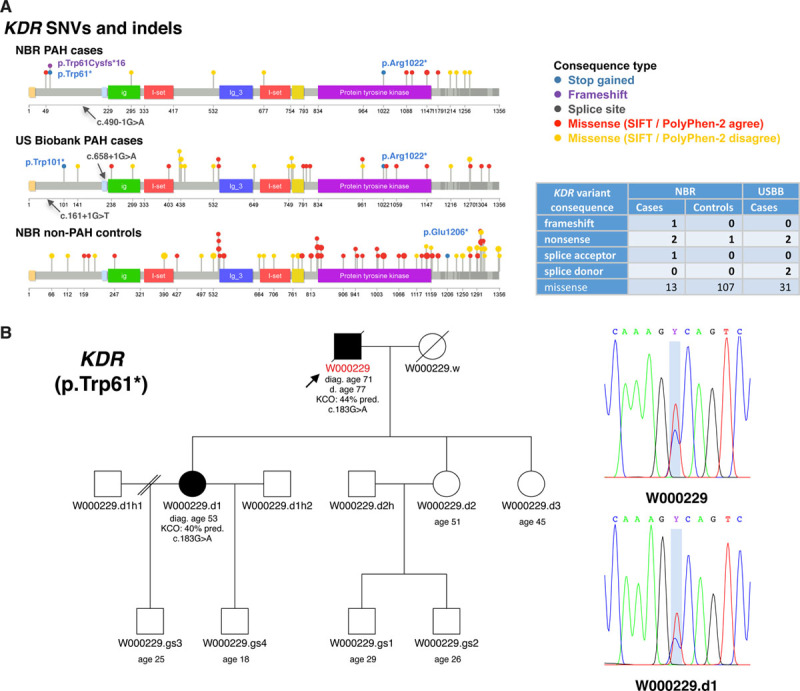
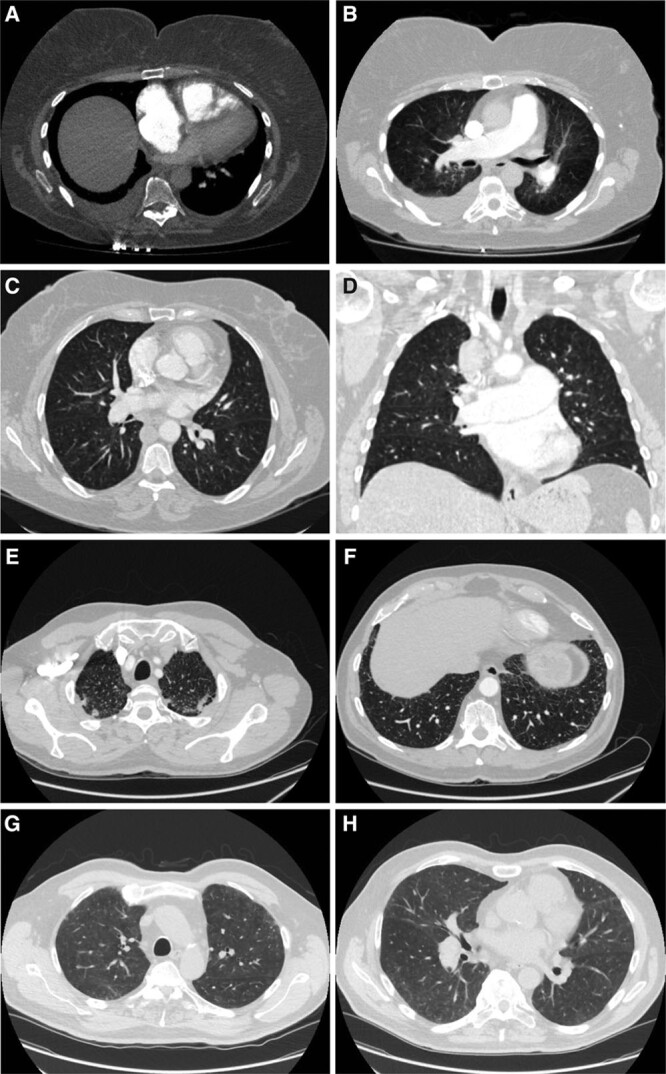
Background - Approximately 25% of patients with pulmonary arterial hypertension (PAH) have been found to harbor rare mutations in disease-causing genes. To identify missing heritability in PAH we integrated deep phenotyping with whole-genome sequencing data using Bayesian statistics. Methods - We analyzed 13,037 participants enrolled in the NIHR BioResource - Rare Diseases (NBR) study, of which 1,148 were recruited to the PAH domain. To test for genetic associations between genes and selected phenotypes of pulmonary hypertension (PH), we used the Bayesian rare-variant association method BeviMed. Results - Heterozygous, high impact, likely loss-of-function variants in the Kinase Insert Domain Receptor (KDR) gene were strongly associated with significantly reduced transfer coefficient for carbon monoxide (KCO, posterior probability (PP)=0.989) and older age at diagnosis (PP=0.912). We also provide evidence for familial segregation of a rare nonsense KDR variant with these phenotypes. On computed tomographic imaging of the lungs, a range of parenchymal abnormalities were observed in the five patients harboring these predicted deleterious variants in KDR. Four additional PAH cases with rare likely loss-of-function variants in KDR were independently identified in the US PAH Biobank cohort with similar phenotypic characteristics. Conclusions - The Bayesian inference approach allowed us to independently validate KDR, which encodes for the Vascular Endothelial Growth Factor Receptor 2 (VEGFR2), as a novel PAH candidate gene. Furthermore, this approach specifically associated high impact likely loss-of-function variants in the genetically constrained gene with distinct phenotypes. These findings provide evidence for KDR being a clinically actionable PAH gene and further support the central role of the vascular endothelium in the pathobiology of PAH.
Keywords: computed tomography.
Conflict of interest statement
Dr Morrell is a Director and Co-founder of Morphogen-IX. Dr Wharton received personal fees from Actelion Pharmaceuticals. Dr Kovacs reports personal fees and nonfinancial support from Actelion Pharmaceuticals, Bayer, GlaxoSmithKline, Merck Sharp & Dohme Corp., Boehringer Ingelheim, Novartis, Chiesi, and Vitalaire outside the submitted work. Dr Penkett declares fees from Actelion Pharmaceuticals and United Therapeutics. Dr Lawrie received support and fees from GlaxoSmithKline and Actelion Pharmaceuticals.
Figures
References
-
- Pullamsetti SS, Savai R, Seeger W, Goncharova EA. Translational advances in the field of pulmonary hypertension. From cancer biology to new pulmonary arterial hypertension therapeutics. Targeting cell growth and proliferation signaling hubs. Am J Respir Crit Care Med. 2017;195:425–437. doi: 10.1164/rccm.201606-1226PP - PMC - PubMed
-
- Lane KB, Machado RD, Pauciulo MW, Thomson JR, Phillips JA, III, Loyd JE, Nichols WC, Trembath RC; International PPH Consortium. Heterozygous germline mutations in BMPR2, encoding a TGF-beta receptor, cause familial primary pulmonary hypertension Nat Genet. 2000;26:81–84. - PubMed
-
- Trembath RC. Mutations in the TGF-beta type 1 receptor, ALK1, in combined primary pulmonary hypertension and hereditary haemorrhagic telangiectasia, implies pathway specificity. J Heart Lung Transplant. 2001;20:175 doi: 10.1016/s1053-2498(00)00352-1 - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
