

A Computer-Aided Drug Design Approach to Predict Marine Drug-Like Leads for SARS-CoV-2 Main Protease Inhibition
- PMID: 33322052
- PMCID: PMC7764804
- DOI: 10.3390/md18120633
A Computer-Aided Drug Design Approach to Predict Marine Drug-Like Leads for SARS-CoV-2 Main Protease Inhibition
Abstract
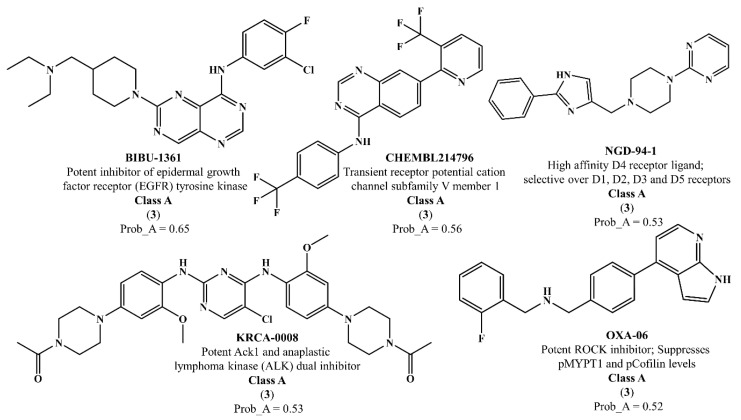
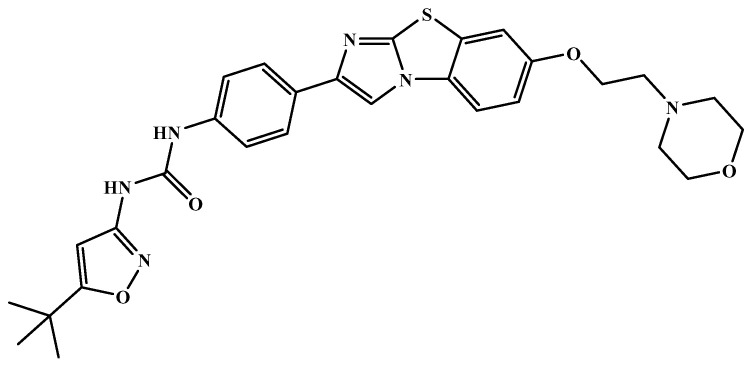
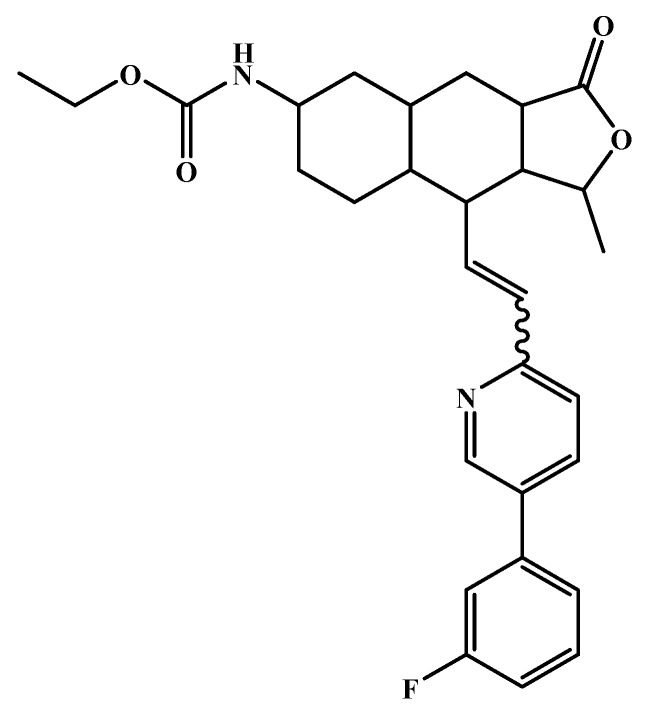
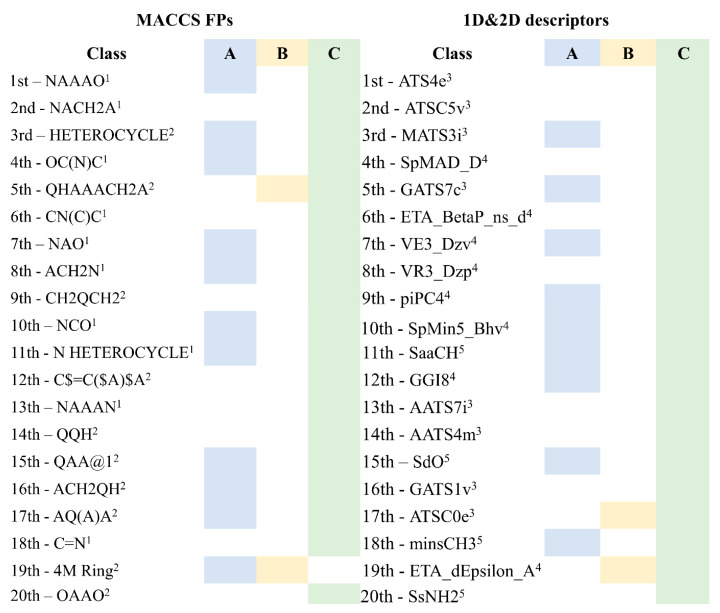
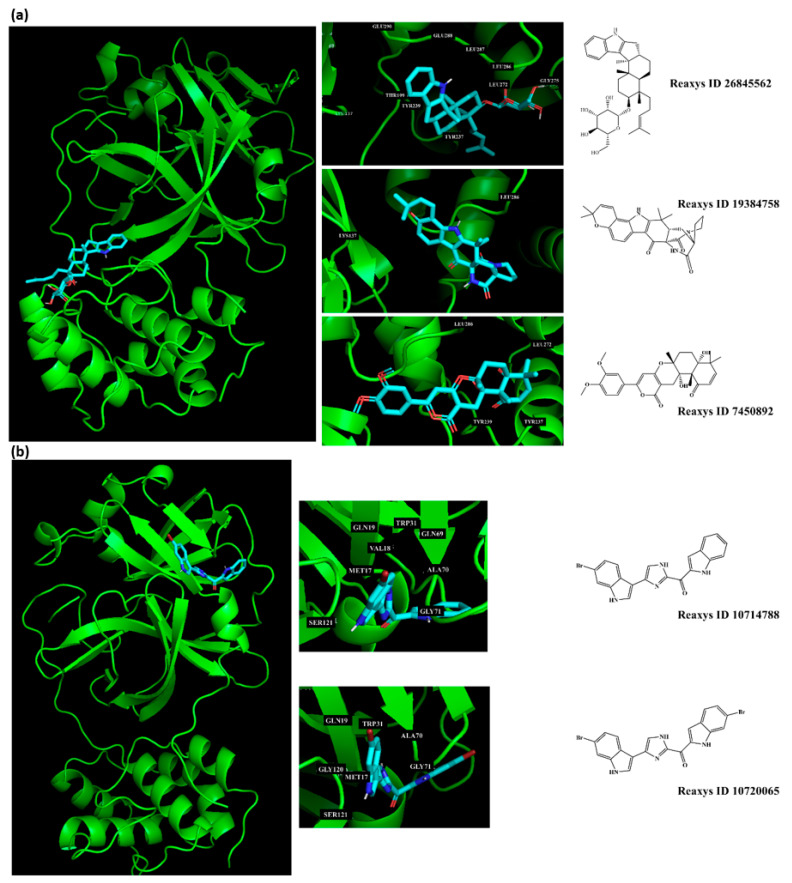
The investigation of marine natural products (MNPs) as key resources for the discovery of drugs to mitigate the COVID-19 pandemic is a developing field. In this work, computer-aided drug design (CADD) approaches comprising ligand- and structure-based methods were explored for predicting SARS-CoV-2 main protease (Mpro) inhibitors. The CADD ligand-based method used a quantitative structure-activity relationship (QSAR) classification model that was built using 5276 organic molecules extracted from the ChEMBL database with SARS-CoV-2 screening data. The best model achieved an overall predictive accuracy of up to 67% for an external and internal validation using test and training sets. Moreover, based on the best QSAR model, a virtual screening campaign was carried out using 11,162 MNPs retrieved from the Reaxys® database, 7 in-house MNPs obtained from marine-derived actinomycetes by the team, and 14 MNPs that are currently in the clinical pipeline. All the MNPs from the virtual screening libraries that were predicted as belonging to class A were selected for the CADD structure-based method. In the CADD structure-based approach, the 494 MNPs selected by the QSAR approach were screened by molecular docking against Mpro enzyme. A list of virtual screening hits comprising fifteen MNPs was assented by establishing several limits in this CADD approach, and five MNPs were proposed as the most promising marine drug-like leads as SARS-CoV-2 Mpro inhibitors, a benzo[f]pyrano[4,3-b]chromene, notoamide I, emindole SB beta-mannoside, and two bromoindole derivatives.
Keywords: actinomycetes; drug discovery; machine learning (ML) techniques; main protease enzyme (Mpro); marine natural products (MNPs); molecular docking; quantitative structure–activity relationship (QSAR); severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2); virtual screening.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
Predicting Antifouling Activity and Acetylcholinesterase Inhibition of Marine-Derived Compounds Using a Computer-Aided Drug Design Approach.Mar Drugs. 2022 Feb 8;20(2):129. doi: 10.3390/md20020129. Mar Drugs. 2022. PMID: 35200658 Free PMC article.
-
Drugs Repurposing Using QSAR, Docking and Molecular Dynamics for Possible Inhibitors of the SARS-CoV-2 Mpro Protease.Molecules. 2020 Nov 6;25(21):5172. doi: 10.3390/molecules25215172. Molecules. 2020. PMID: 33172092 Free PMC article.
-
Investigating the antiviral therapeutic potentialities of marine polycyclic lamellarin pyrrole alkaloids as promising inhibitors for SARS-CoV-2 and Zika main proteases (Mpro).J Biomol Struct Dyn. 2024 May;42(8):3983-4001. doi: 10.1080/07391102.2023.2217513. Epub 2023 May 26. J Biomol Struct Dyn. 2024. PMID: 37232419
-
SARS-CoV-2 Mpro: A Potential Target for Peptidomimetics and Small-Molecule Inhibitors.Biomolecules. 2021 Apr 19;11(4):607. doi: 10.3390/biom11040607. Biomolecules. 2021. PMID: 33921886 Free PMC article. Review.
-
Docking Paradigm in Drug Design.Curr Top Med Chem. 2021;21(6):507-546. doi: 10.2174/1568026620666201207095626. Curr Top Med Chem. 2021. PMID: 33292135 Review.
Cited by
-
Computational repurposing of asthma drugs as potential inhibitors of SARS-CoV-2 Mpro.New Microbes New Infect. 2022 Apr-May;47:100979. doi: 10.1016/j.nmni.2022.100979. Epub 2022 Apr 11. New Microbes New Infect. 2022. PMID: 35433012 Free PMC article. No abstract available.
-
Implication of in silico studies in the search for novel inhibitors against SARS-CoV-2.Arch Pharm (Weinheim). 2022 May;355(5):e2100360. doi: 10.1002/ardp.202100360. Epub 2022 Mar 4. Arch Pharm (Weinheim). 2022. PMID: 35244237 Free PMC article. Review.
-
Advanced Methods for Natural Products Discovery: Bioactivity Screening, Dereplication, Metabolomics Profiling, Genomic Sequencing, Databases and Informatic Tools, and Structure Elucidation.Mar Drugs. 2023 May 19;21(5):308. doi: 10.3390/md21050308. Mar Drugs. 2023. PMID: 37233502 Free PMC article. Review.
-
Marine natural products and human immunity: novel biomedical resources for anti-infection of SARS-CoV-2 and related cardiovascular disease.Nat Prod Bioprospect. 2024 Jan 29;14(1):12. doi: 10.1007/s13659-024-00432-4. Nat Prod Bioprospect. 2024. PMID: 38282092 Free PMC article. Review.
-
Computational Identification of Potential Anti-Inflammatory Natural Compounds Targeting the p38 Mitogen-Activated Protein Kinase (MAPK): Implications for COVID-19-Induced Cytokine Storm.Biomolecules. 2021 Apr 29;11(5):653. doi: 10.3390/biom11050653. Biomolecules. 2021. PMID: 33946644 Free PMC article.
References
-
- Coronavirus Resource Center Global Tracking. [(accessed on 9 December 2020)]; Available online: https://coronavirus.jhu.edu/map.html.
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
