

FIREWORKS: a bottom-up approach to integrative coessentiality network analysis
- PMID: 33328249
- PMCID: PMC7756899
- DOI: 10.26508/lsa.202000882
FIREWORKS: a bottom-up approach to integrative coessentiality network analysis
Abstract
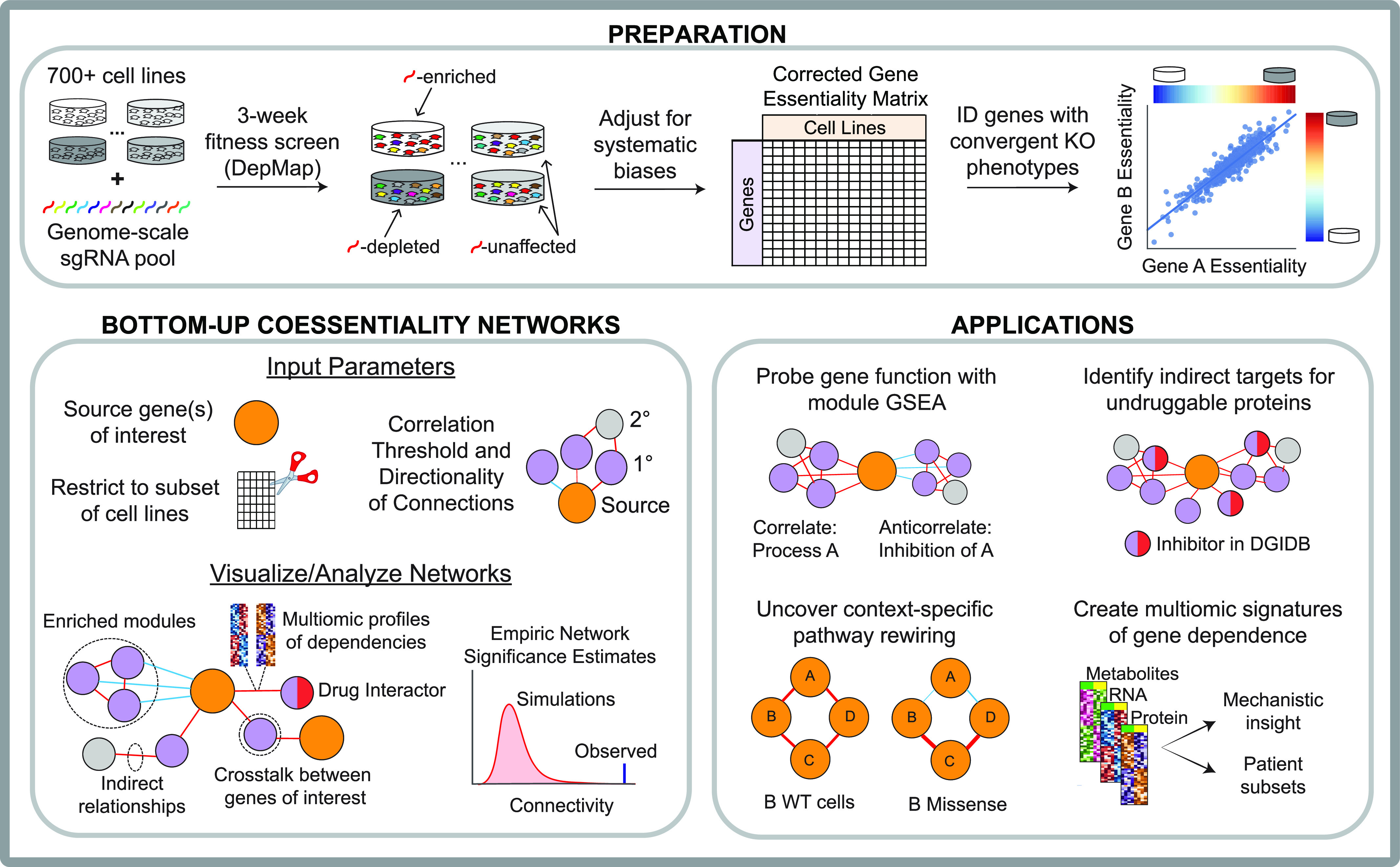
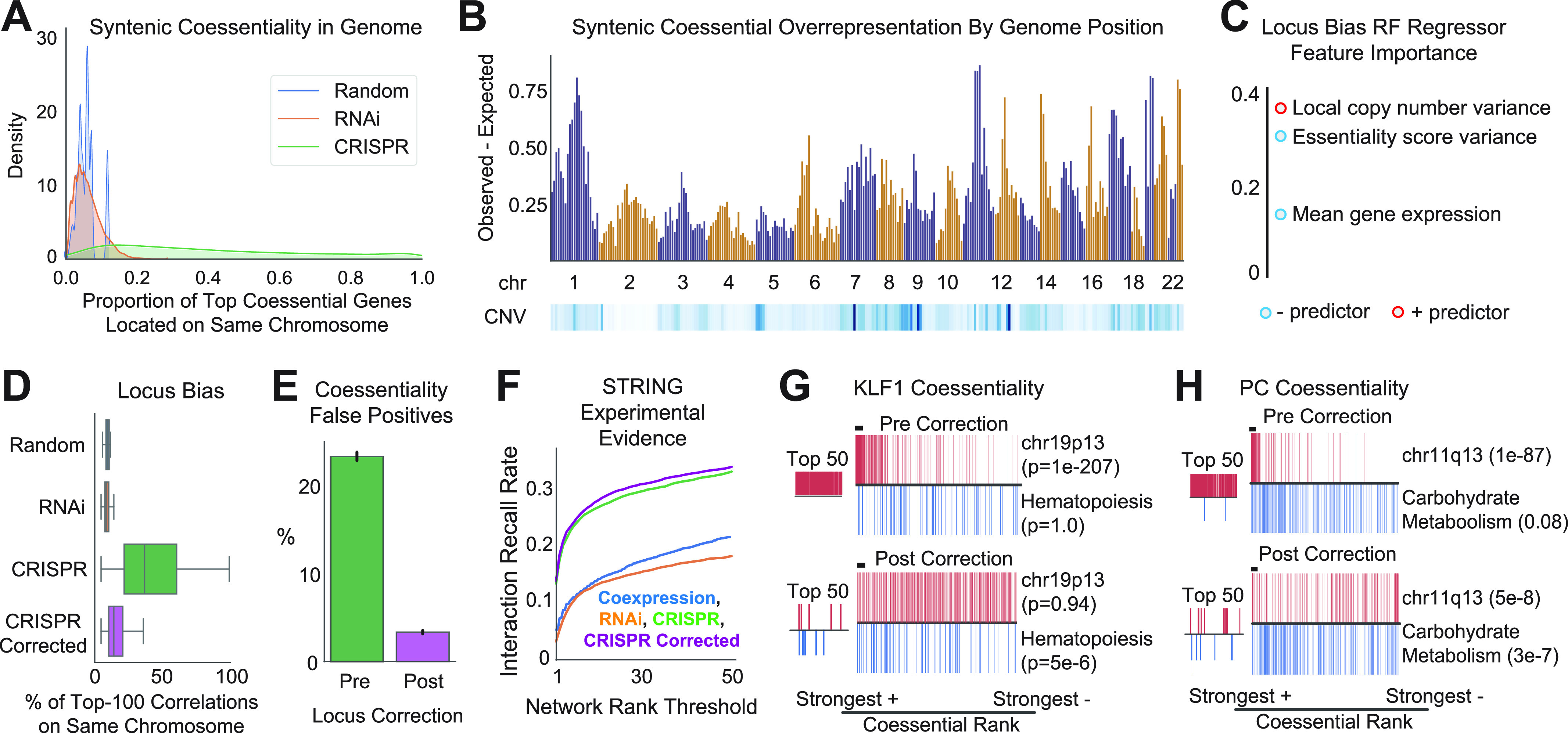
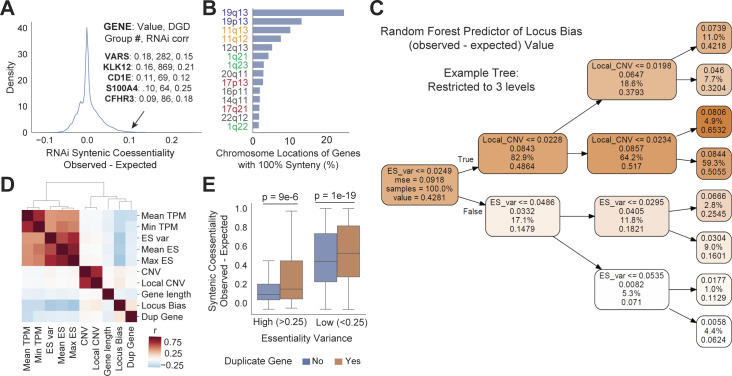
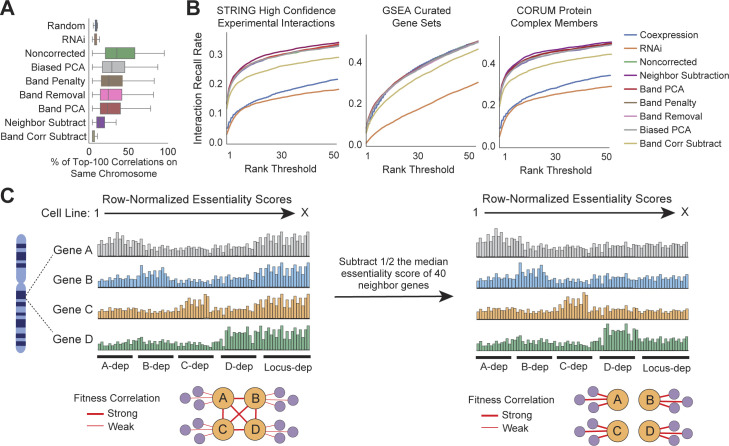
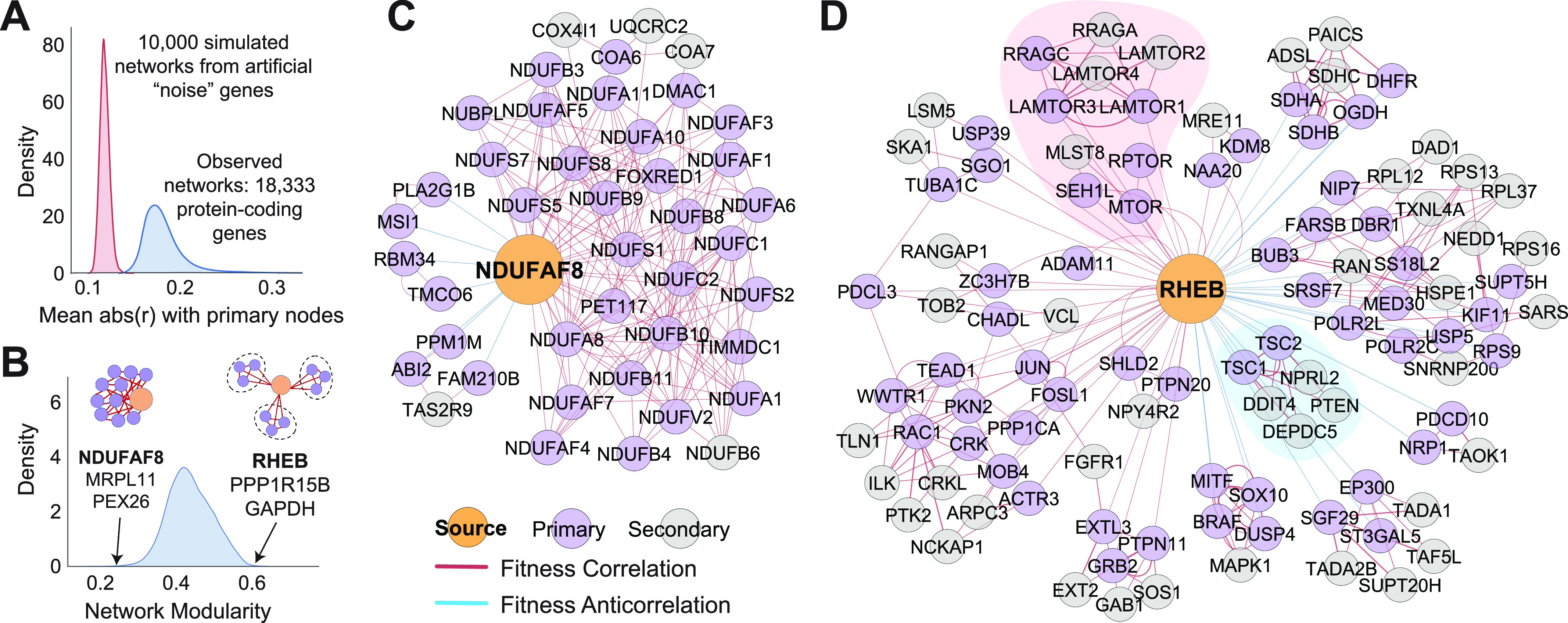
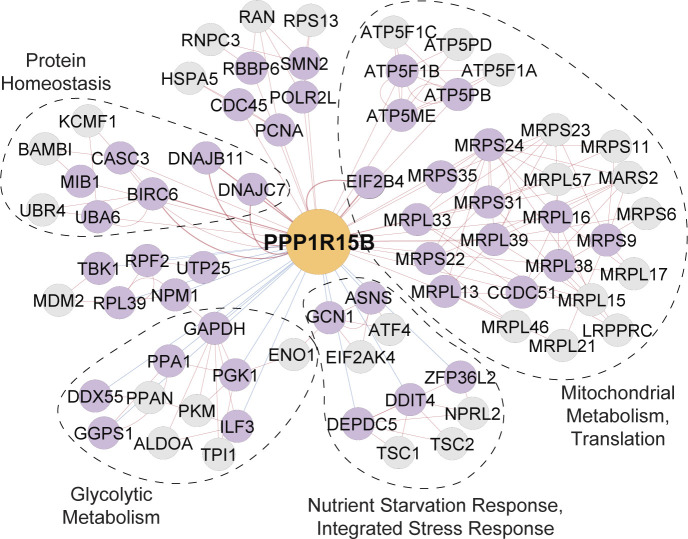
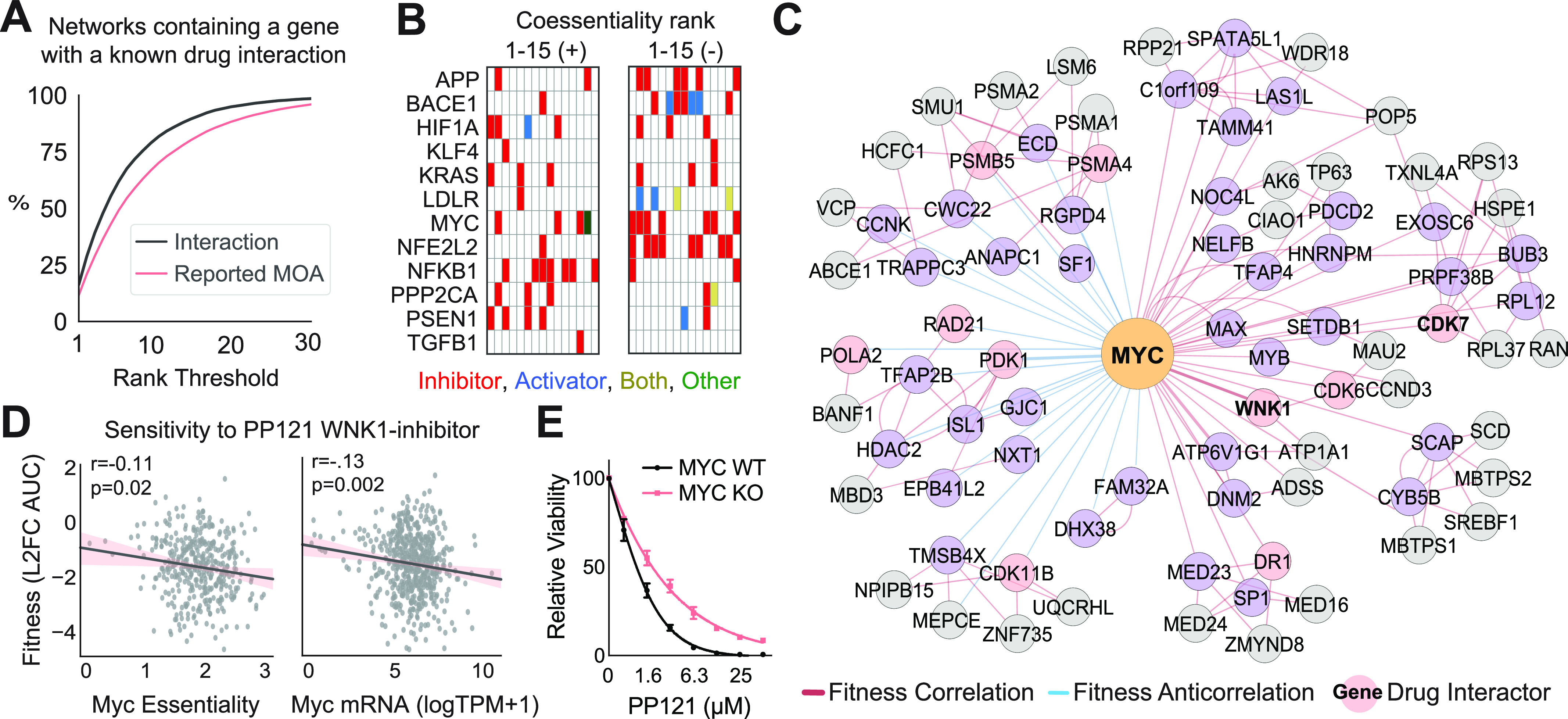
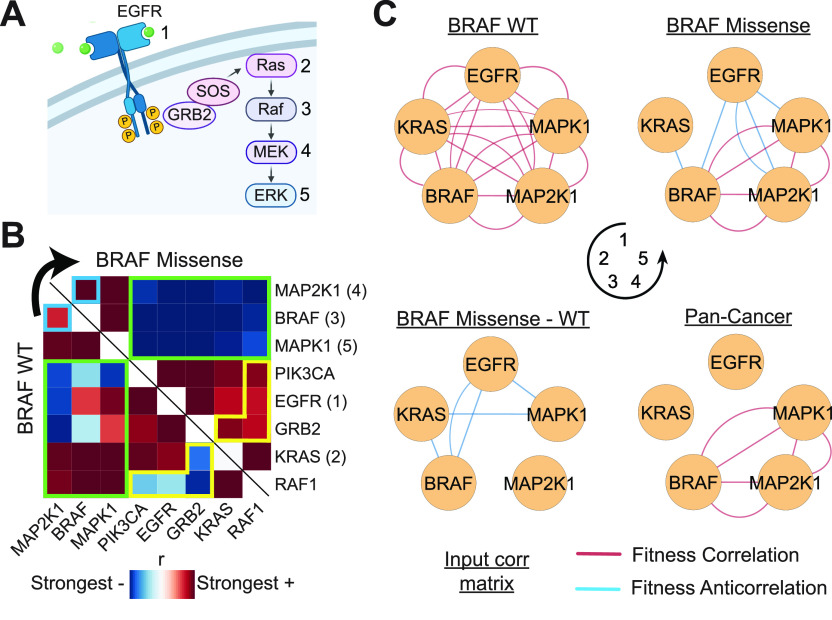
Genetic coessentiality analysis, a computational approach which identifies genes sharing a common effect on cell fitness across large-scale screening datasets, has emerged as a powerful tool to identify functional relationships between human genes. However, widespread implementation of coessentiality to study individual genes and pathways is limited by systematic biases in existing coessentiality approaches and accessibility barriers for investigators without computational expertise. We created FIREWORKS, a method and interactive tool for the construction and statistical analysis of coessentiality networks centered around gene(s) provided by the user. FIREWORKS incorporates a novel bias reduction approach to reduce false discoveries, enables restriction of coessentiality analyses to custom subsets of cell lines, and integrates multiomic and drug-gene interaction datasets to investigate and target contextual gene essentiality. We demonstrate the broad utility of FIREWORKS through case vignettes investigating gene function and specialization, indirect therapeutic targeting of "undruggable" proteins, and context-specific rewiring of genetic networks.
© 2020 Amici et al.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures
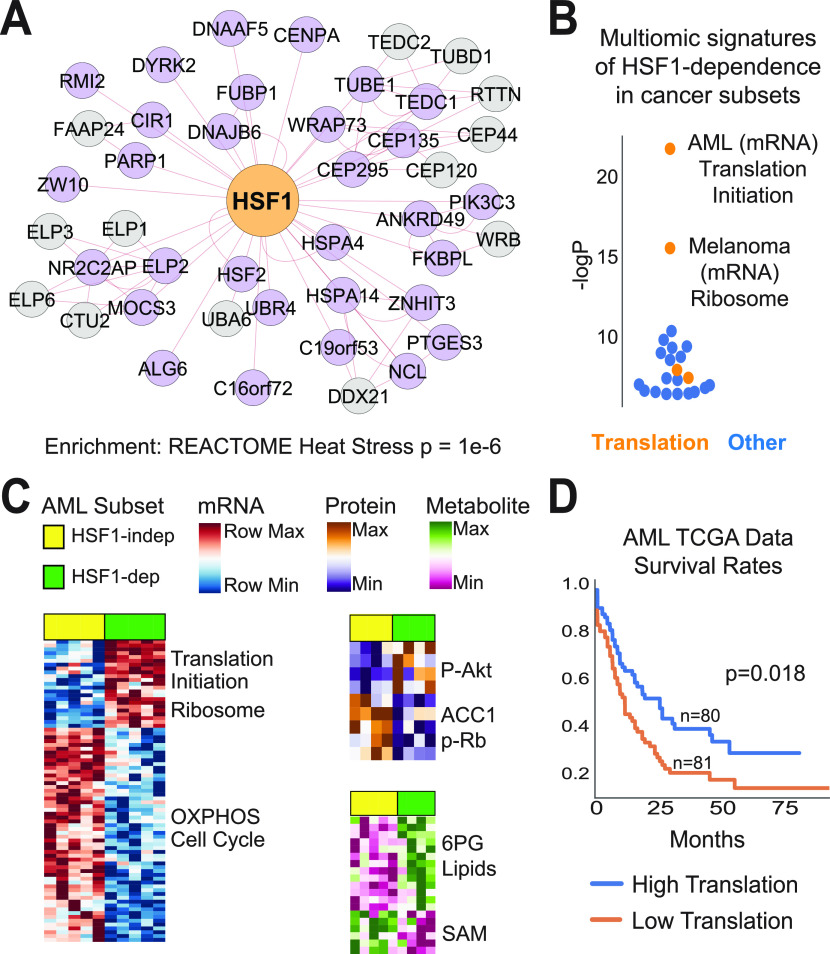
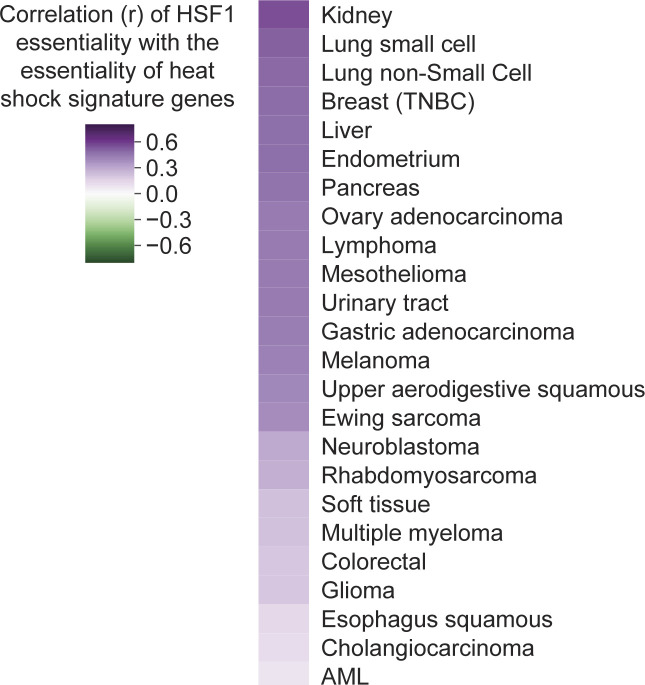
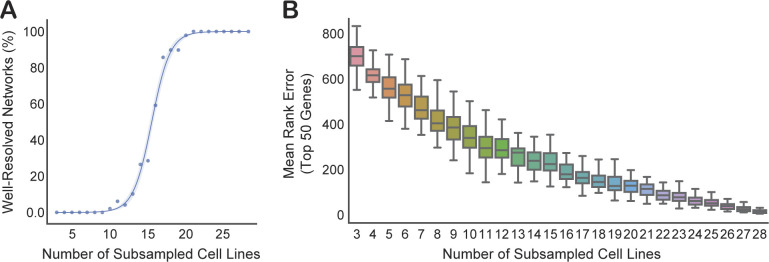
References
-
- Bayraktar EC, La K, Karpman K, Unlu G, Ozerdem C, Ritter DJ, Alwaseem H, Molina H, Hoffmann H-H, Millner A, et al. (2020) Metabolic coessentiality mapping identifies C12orf49 as a regulator of SREBP processing and cholesterol metabolism. Nat Metab 2: 487–498. 10.1038/s42255-020-0206-9 - DOI - PMC - PubMed
-
- Blondel VD, Guillaume JL, Lambiotte R, Lefebvre E (2008) Fast unfolding of communities in large networks. J Stat Mech Theor Exp 2008: P10008 10.1088/1742-5468/2008/10/p10008:P100008 - DOI
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources