

Measuring DNA mechanics on the genome scale
- PMID: 33328628
- PMCID: PMC7855230
- DOI: 10.1038/s41586-020-03052-3
Measuring DNA mechanics on the genome scale
Abstract
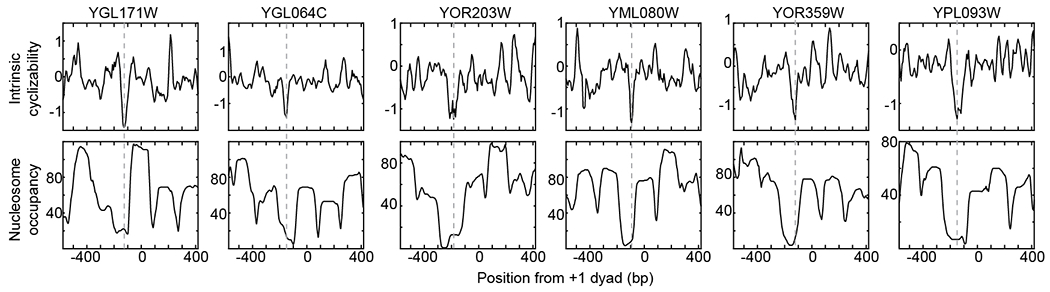
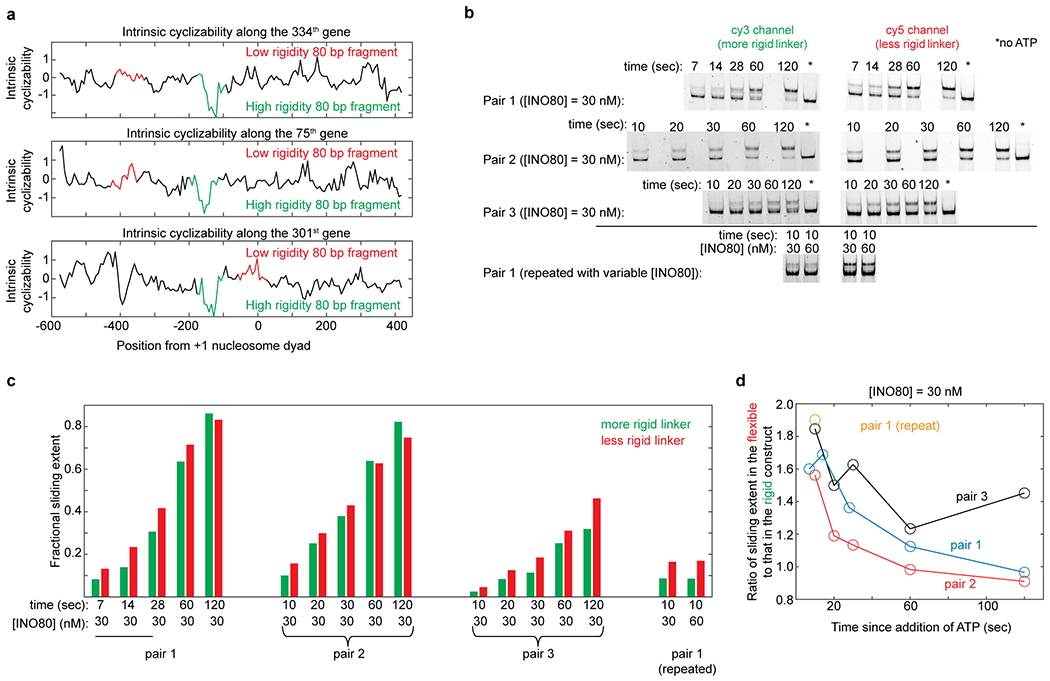
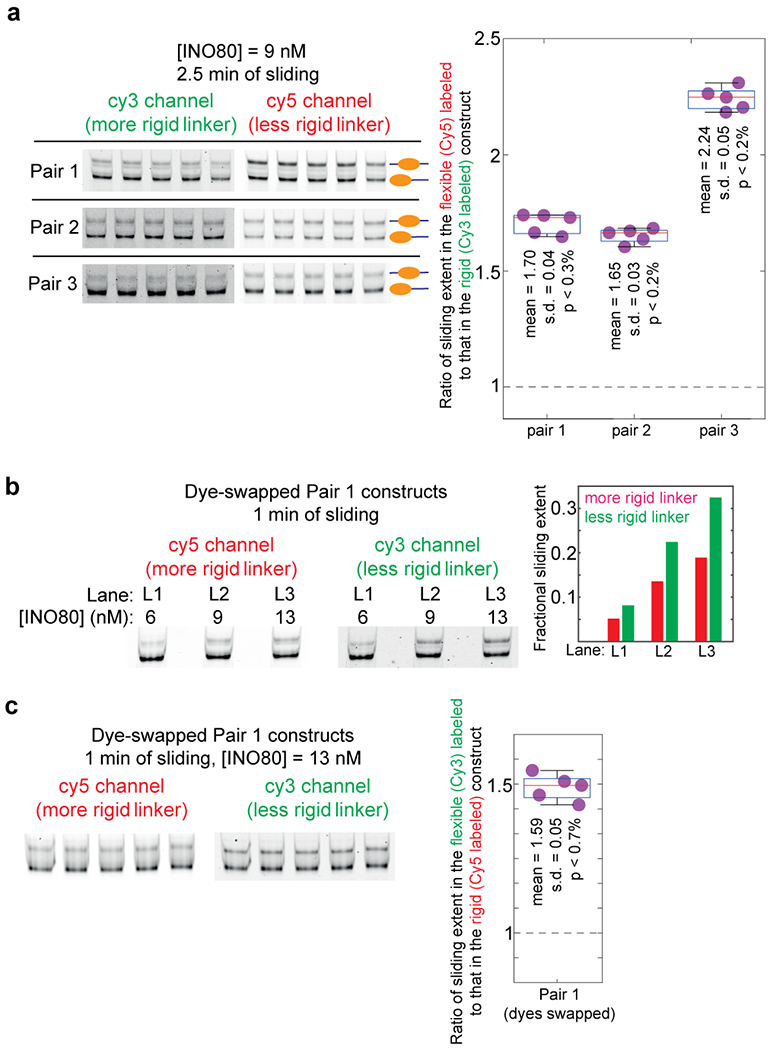
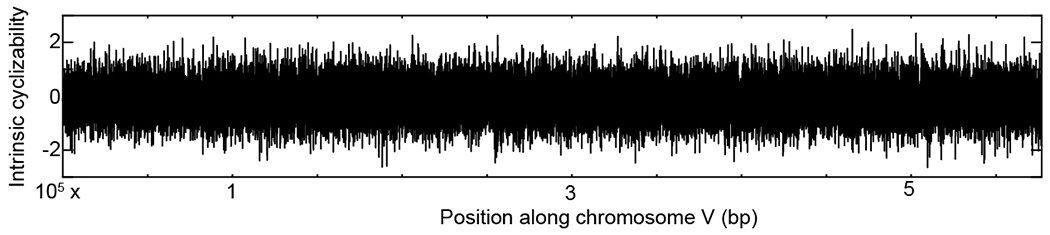
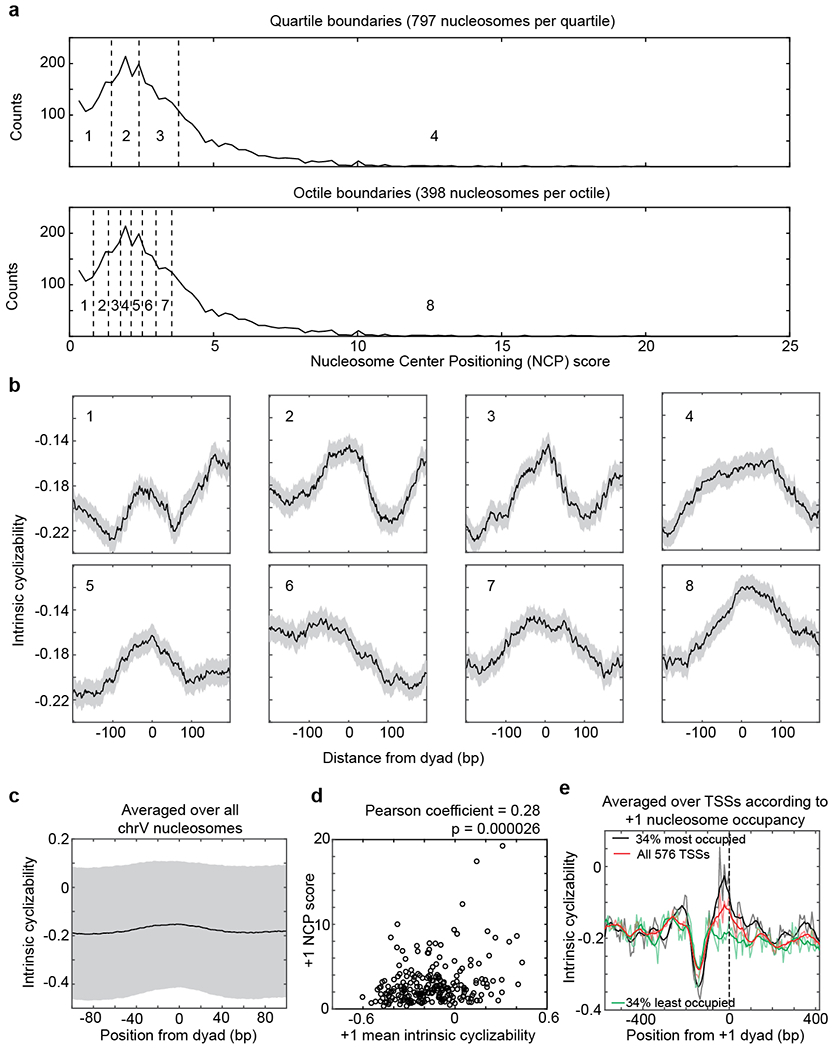
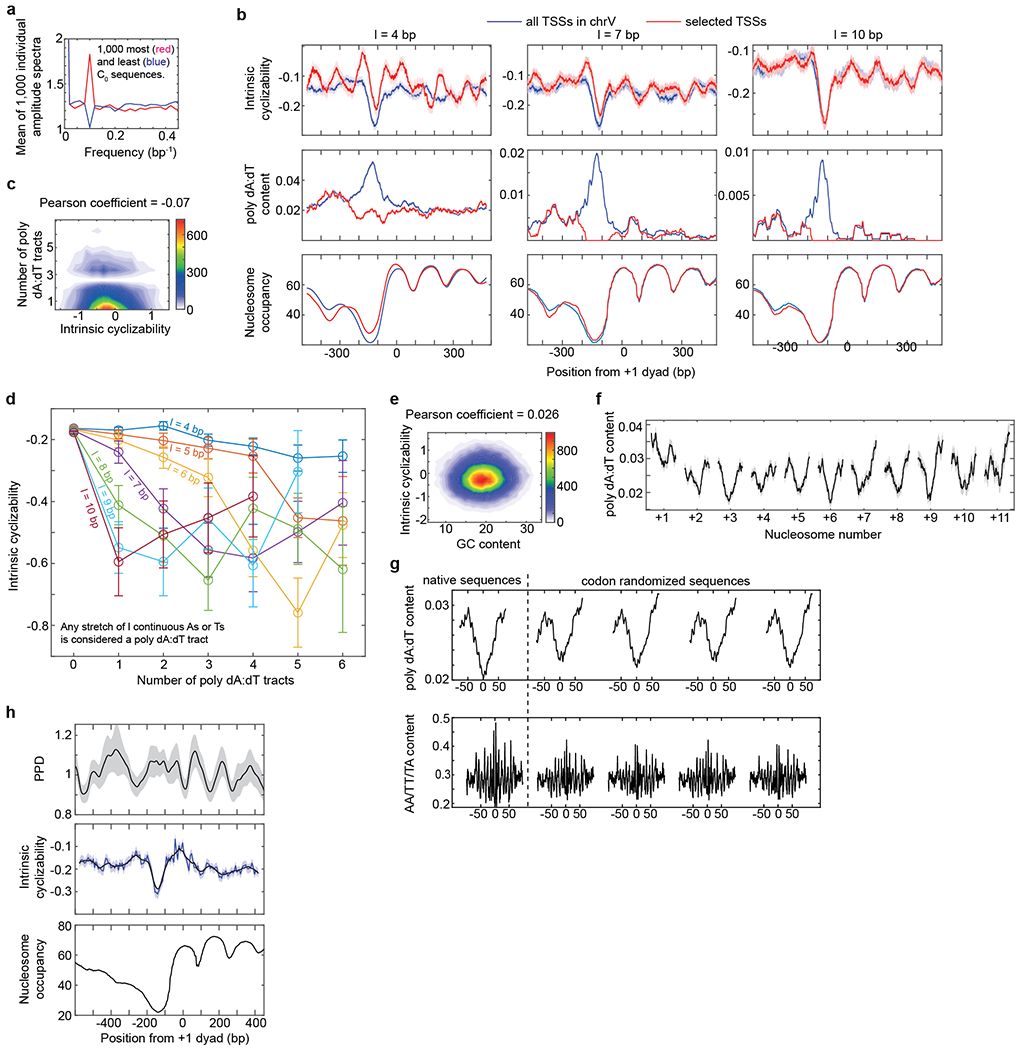
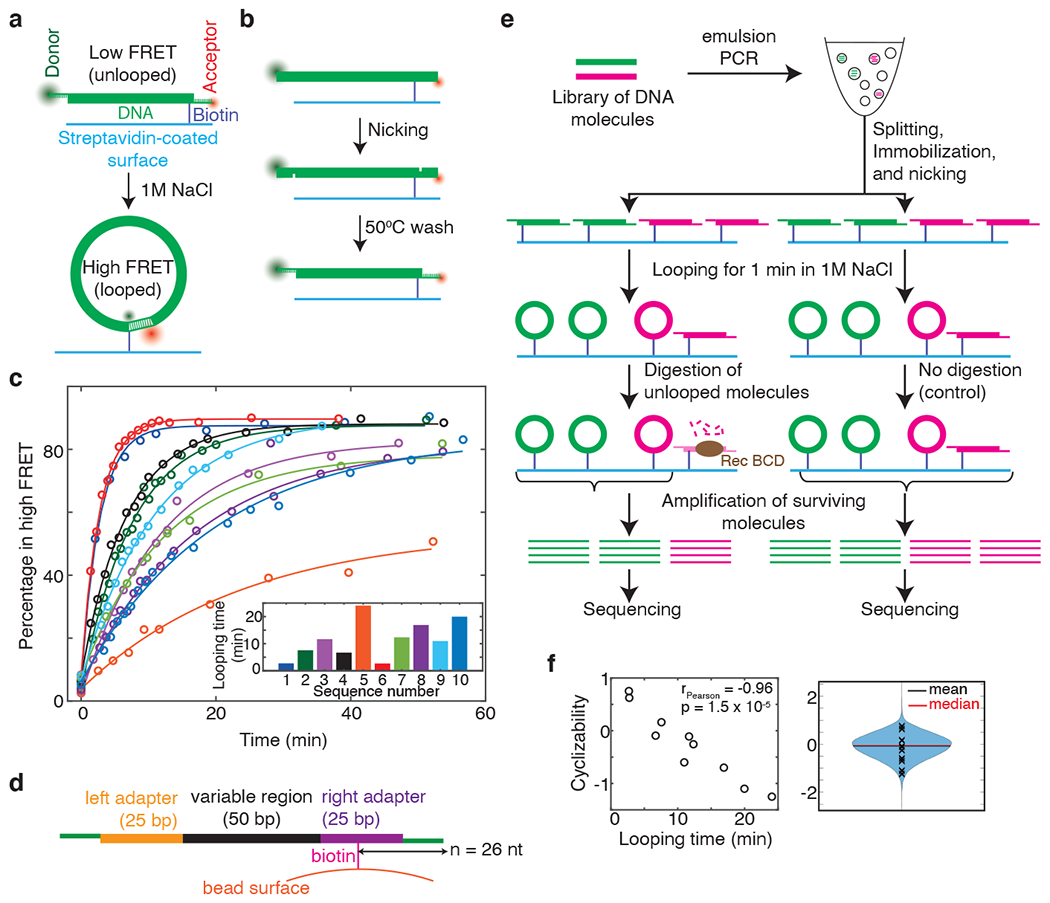
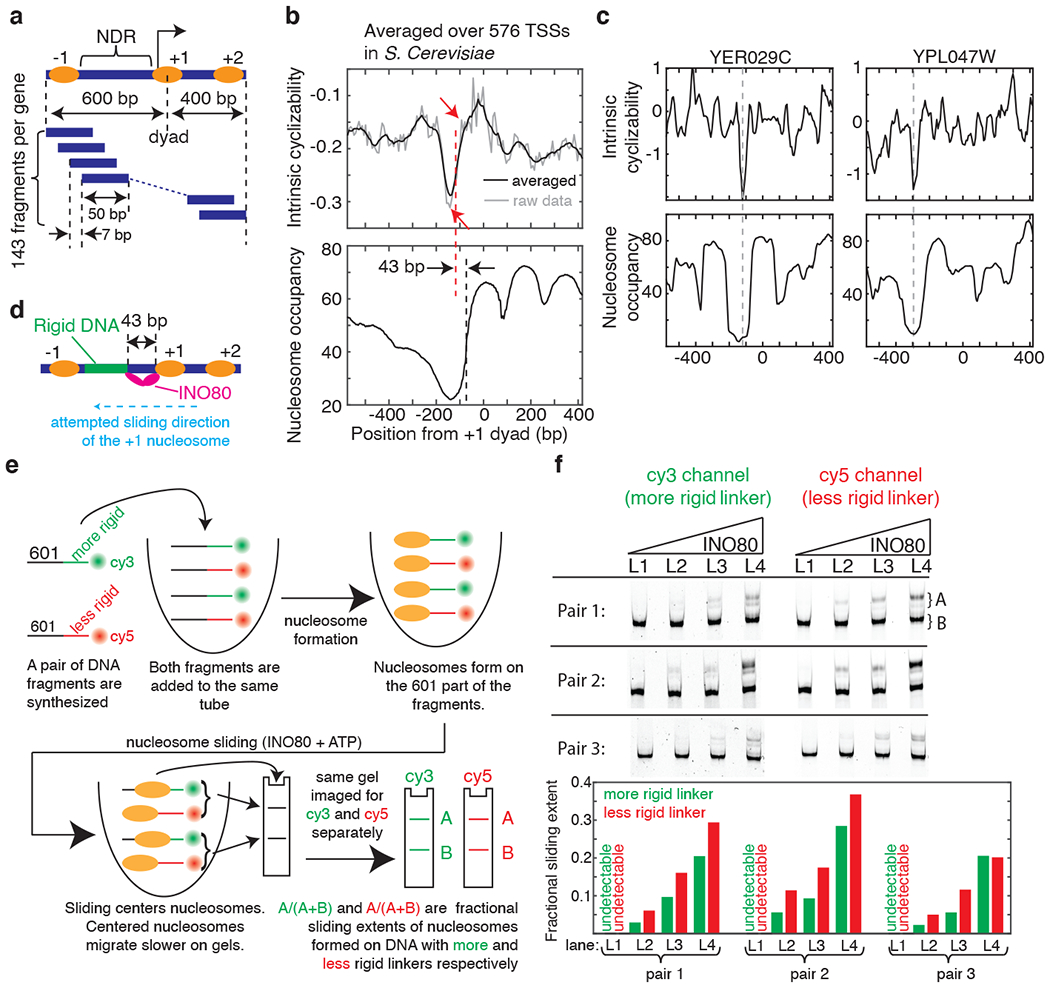
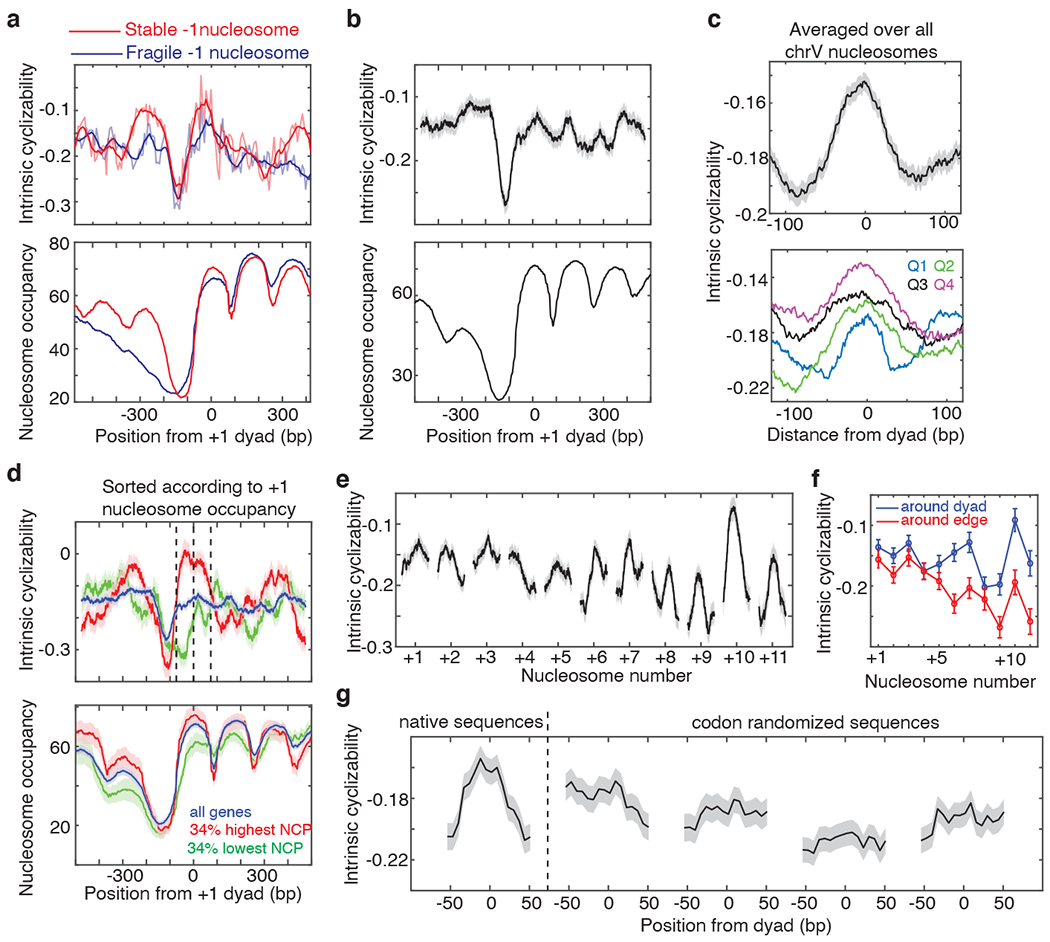
Mechanical deformations of DNA such as bending are ubiquitous and have been implicated in diverse cellular functions1. However, the lack of high-throughput tools to measure the mechanical properties of DNA has limited our understanding of how DNA mechanics influence chromatin transactions across the genome. Here we develop 'loop-seq'-a high-throughput assay to measure the propensity for DNA looping-and determine the intrinsic cyclizabilities of 270,806 50-base-pair DNA fragments that span Saccharomyces cerevisiae chromosome V, other genomic regions, and random sequences. We found sequence-encoded regions of unusually low bendability within nucleosome-depleted regions upstream of transcription start sites (TSSs). Low bendability of linker DNA inhibits nucleosome sliding into the linker by the chromatin remodeller INO80, which explains how INO80 can define nucleosome-depleted regions in the absence of other factors2. Chromosome-wide, nucleosomes were characterized by high DNA bendability near dyads and low bendability near linkers. This contrast increases for deeper gene-body nucleosomes but disappears after random substitution of synonymous codons, which suggests that the evolution of codon choice has been influenced by DNA mechanics around gene-body nucleosomes. Furthermore, we show that local DNA mechanics affect transcription through TSS-proximal nucleosomes. Overall, this genome-scale map of DNA mechanics indicates a 'mechanical code' with broad functional implications.
Conflict of interest statement
Figures

Comment in
-
Sequencing DNA bendability.Nat Methods. 2021 Feb;18(2):121. doi: 10.1038/s41592-021-01070-1. Nat Methods. 2021. PMID: 33542507 No abstract available.
-
Uncovering the Mechanical Code of DNA Using 'Loop-seq'.Trends Genet. 2021 Jun;37(6):494-495. doi: 10.1016/j.tig.2021.02.004. Epub 2021 Feb 20. Trends Genet. 2021. PMID: 33622494
-
More Than Just Letters and Chemistry: Genomics Goes Mechanics.Trends Biochem Sci. 2021 Jun;46(6):431-432. doi: 10.1016/j.tibs.2021.03.004. Epub 2021 Apr 12. Trends Biochem Sci. 2021. PMID: 33858745
References
-
- Cloutier TE & Widom J Spontaneous sharp bending of double-stranded DNA. Mol. Cell 14, 355–362 (2004). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
