

Analysis of Indian SARS-CoV-2 Genomes Reveals Prevalence of D614G Mutation in Spike Protein Predicting an Increase in Interaction With TMPRSS2 and Virus Infectivity
- PMID: 33329480
- PMCID: PMC7732478
- DOI: 10.3389/fmicb.2020.594928
Analysis of Indian SARS-CoV-2 Genomes Reveals Prevalence of D614G Mutation in Spike Protein Predicting an Increase in Interaction With TMPRSS2 and Virus Infectivity
Abstract
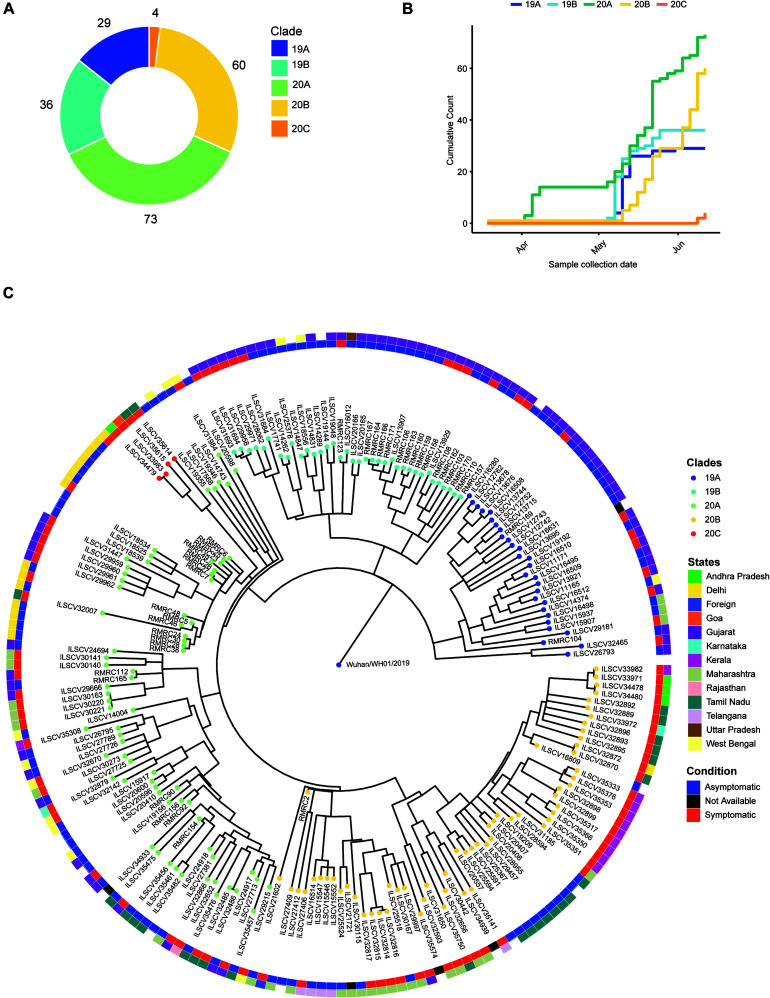
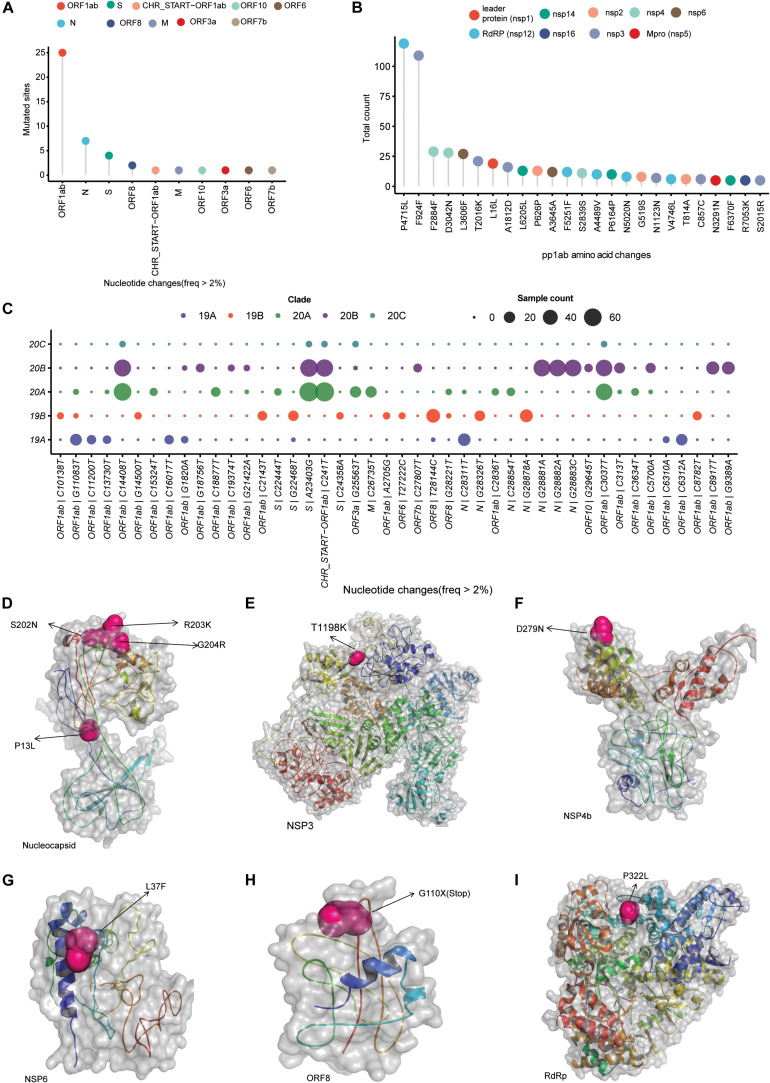
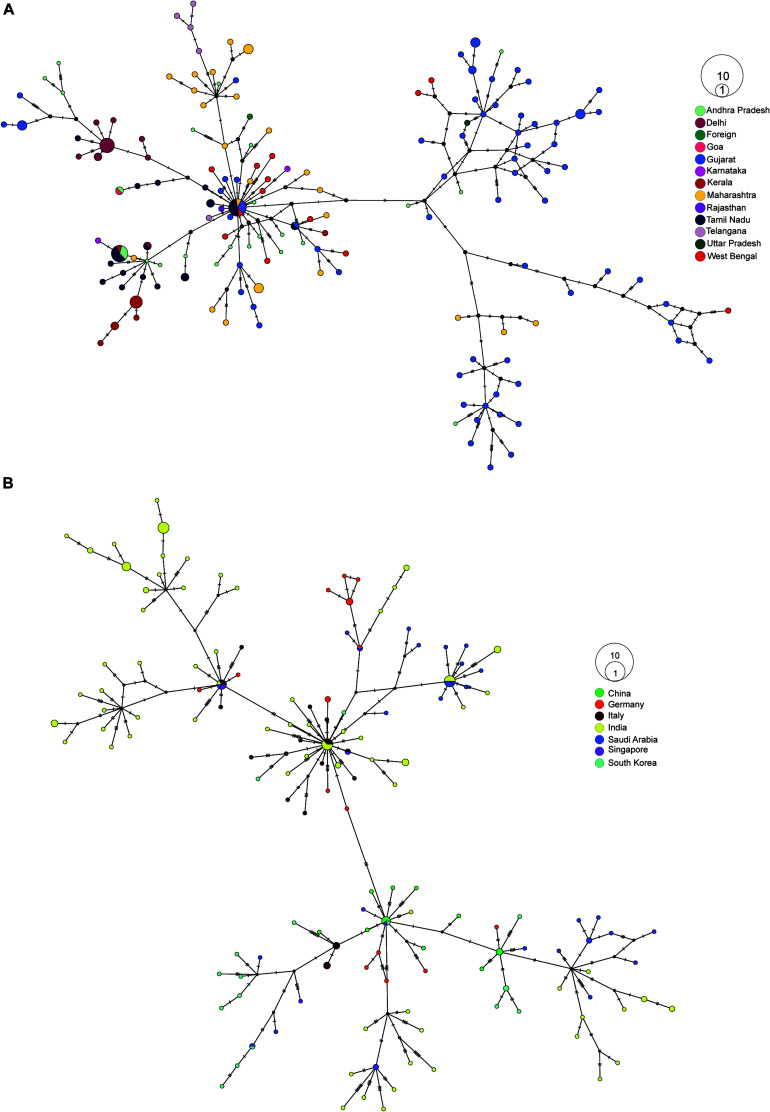
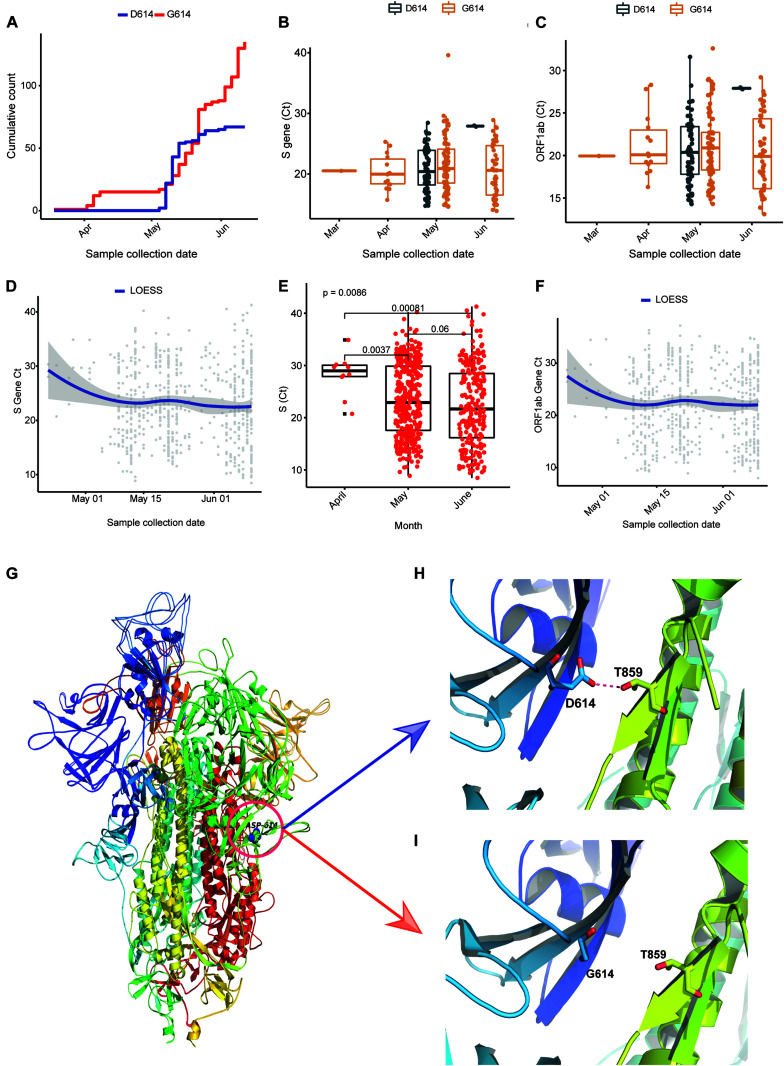
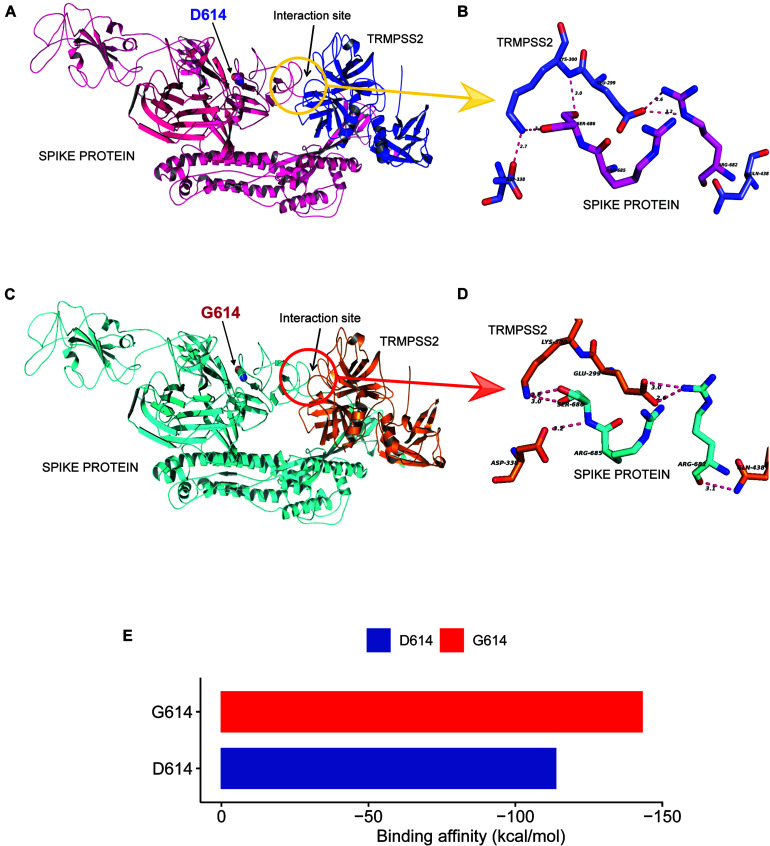
Coronavirus disease 2019 (COVID-19), caused by the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) virus, has emerged as a global pandemic worldwide. In this study, we used ARTIC primers-based amplicon sequencing to profile 225 SARS-CoV-2 genomes from India. Phylogenetic analysis of 202 high-quality assemblies identified the presence of all the five reported clades 19A, 19B, 20A, 20B, and 20C in the population. The analyses revealed Europe and Southeast Asia as two major routes for introduction of the disease in India followed by local transmission. Interestingly, the19B clade was found to be more prevalent in our sequenced genomes (17%) compared to other genomes reported so far from India. Haplotype network analysis showed evolution of 19A and 19B clades in parallel from predominantly Gujarat state in India, suggesting it to be one of the major routes of disease transmission in India during the months of March and April, whereas 20B and 20C appeared to evolve from 20A. At the same time, 20A and 20B clades depicted prevalence of four common mutations 241 C > T in 5' UTR, P4715L, F942F along with D614G in the Spike protein. D614G mutation has been reported to increase virus shedding and infectivity. Our molecular modeling and docking analysis identified that D614G mutation resulted in enhanced affinity of Spike S1-S2 hinge region with TMPRSS2 protease, possibly the reason for increased shedding of S1 domain in G614 as compared to D614. Moreover, we also observed an increased concordance of G614 mutation with the viral load, as evident from decreased Ct value of Spike and the ORF1ab gene.
Keywords: COVID-19; D614G; India; SARS-CoV-2; phylogeny; protein-protein interaction; viral RNA sequencing.
Copyright © 2020 Raghav, Ghosh, Turuk, Kumar, Jha, Madhulika, Priyadarshini, Biswas, Shyamli, Singh, Singh, Singh, Datey, Avula, Smita, Sabat, Bhattacharya, Kshatri, Vasudevan, Suryawanshi, Dash, Senapati, Beuria, Swain, Chattopadhyay, Syed, Dixit, Prasad, Odisha COVID-19 Study Group, ILS COVID-19 Team, Pati, and Parida.
Figures
References
-
- Andrews S. (2010). FastQC: A Quality Control Tool for High Throughput Sequence Data [Online]. Available online at: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/
-
- Cingolani P., Platts A., Wang le L., Coon M., Nguyen T., Wang L., et al. (2012). A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 6 80–92. 10.4161/fly.19695 - DOI - PMC - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous