

Cascabel: A Scalable and Versatile Amplicon Sequence Data Analysis Pipeline Delivering Reproducible and Documented Results
- PMID: 33329686
- PMCID: PMC7718033
- DOI: 10.3389/fgene.2020.489357
Cascabel: A Scalable and Versatile Amplicon Sequence Data Analysis Pipeline Delivering Reproducible and Documented Results
Abstract
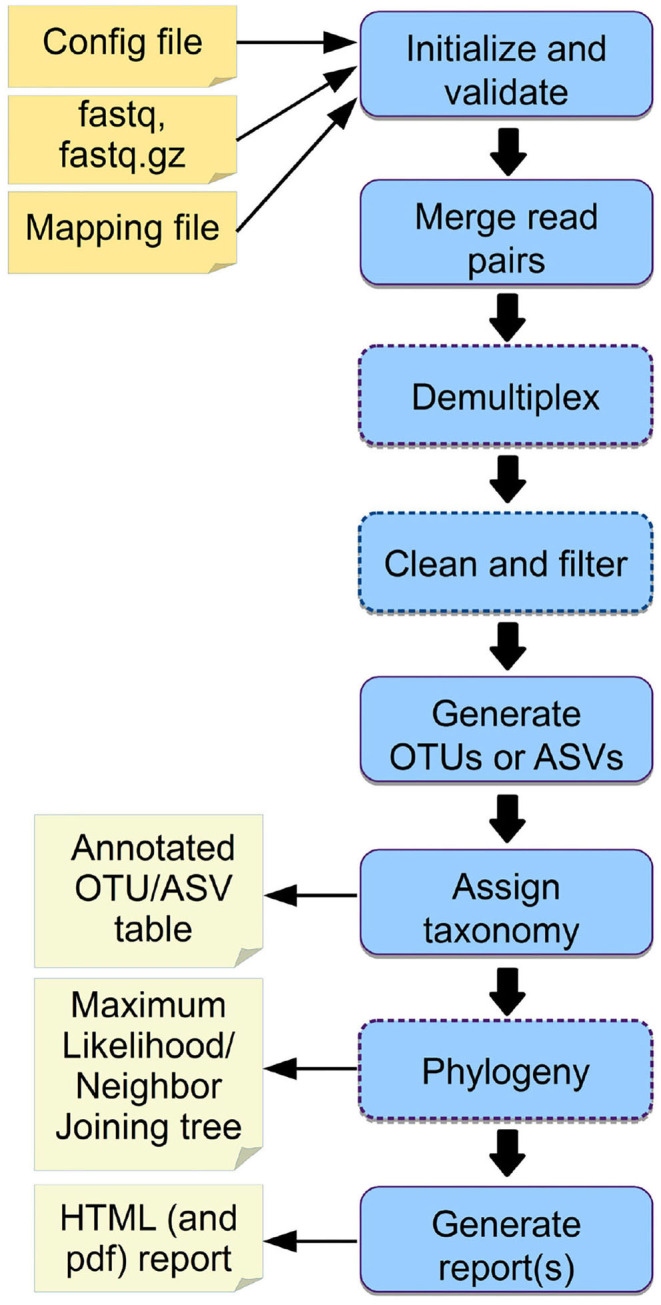
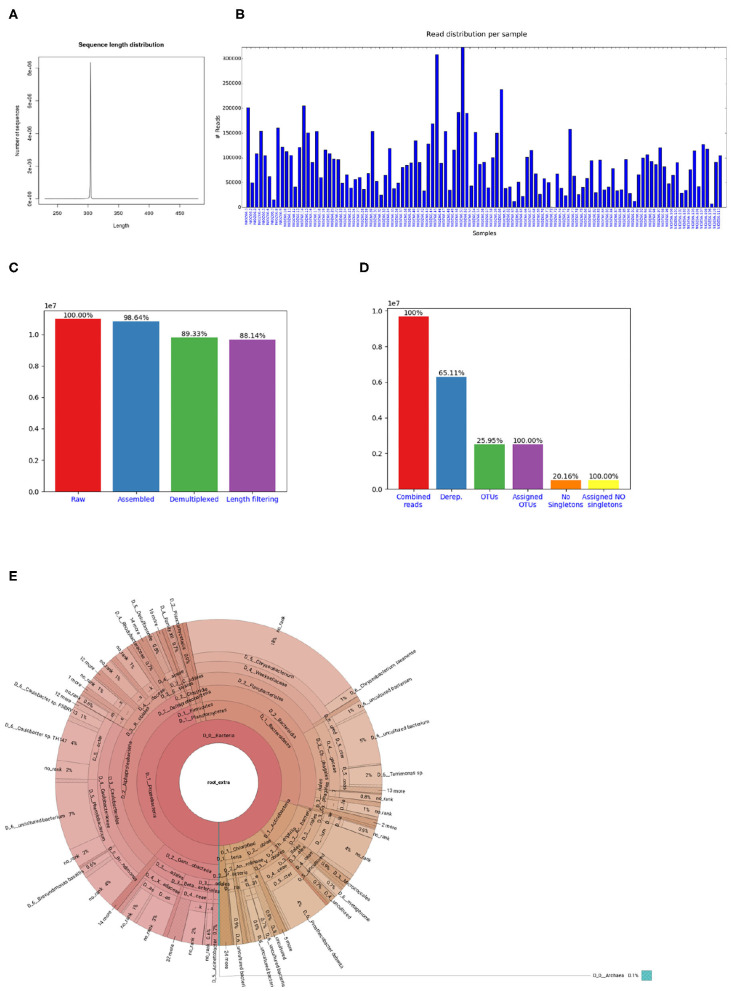
Marker gene sequencing of the rRNA operon (16S, 18S, ITS) or cytochrome c oxidase I (CO1) is a popular means to assess microbial communities of the environment, microbiomes associated with plants and animals, as well as communities of multicellular organisms via environmental DNA sequencing. Since this technique is based on sequencing a single gene, or even only parts of a single gene rather than the entire genome, the number of reads needed per sample to assess the microbial community structure is lower than that required for metagenome sequencing. This makes marker gene sequencing affordable to nearly any laboratory. Despite the relative ease and cost-efficiency of data generation, analyzing the resulting sequence data requires computational skills that may go beyond the standard repertoire of a current molecular biologist/ecologist. We have developed Cascabel, a scalable, flexible, and easy-to-use amplicon sequence data analysis pipeline, which uses Snakemake and a combination of existing and newly developed solutions for its computational steps. Cascabel takes the raw data as input and delivers a table of operational taxonomic units (OTUs) or Amplicon Sequence Variants (ASVs) in BIOM and text format and representative sequences. Cascabel is a highly versatile software that allows users to customize several steps of the pipeline, such as selecting from a set of OTU clustering methods or performing ASV analysis. In addition, we designed Cascabel to run in any linux/unix computing environment from desktop computers to computing servers making use of parallel processing if possible. The analyses and results are fully reproducible and documented in an HTML and optional pdf report. Cascabel is freely available at Github: https://github.com/AlejandroAb/CASCABEL.
Keywords: 16S/18S rRNA gene; Illumina; amplicon sequencing; community profiling; microbiome; pipeline; snakemake.
Copyright © 2020 Abdala Asbun, Besseling, Balzano, van Bleijswijk, Witte, Villanueva and Engelmann.
Figures

References
-
- Andrews S. (2010). FastQC: A Quality Control Tool for High Throughput Sequence Data. Available online at: http://www.bioinformatics.babraham.ac.uk/projects/fastqc
LinkOut - more resources
Full Text Sources