

Defective ALC1 nucleosome remodeling confers PARPi sensitization and synthetic lethality with HRD
- PMID: 33333017
- PMCID: PMC7895907
- DOI: 10.1016/j.molcel.2020.12.006
Defective ALC1 nucleosome remodeling confers PARPi sensitization and synthetic lethality with HRD
Abstract
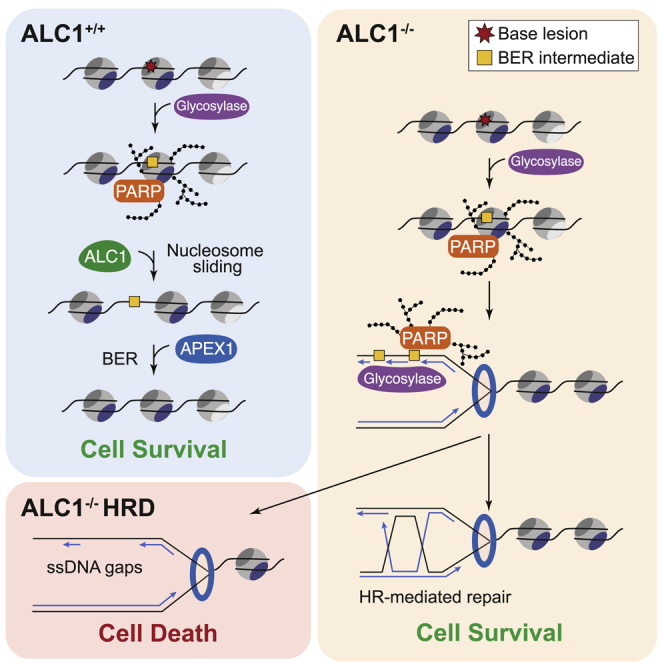
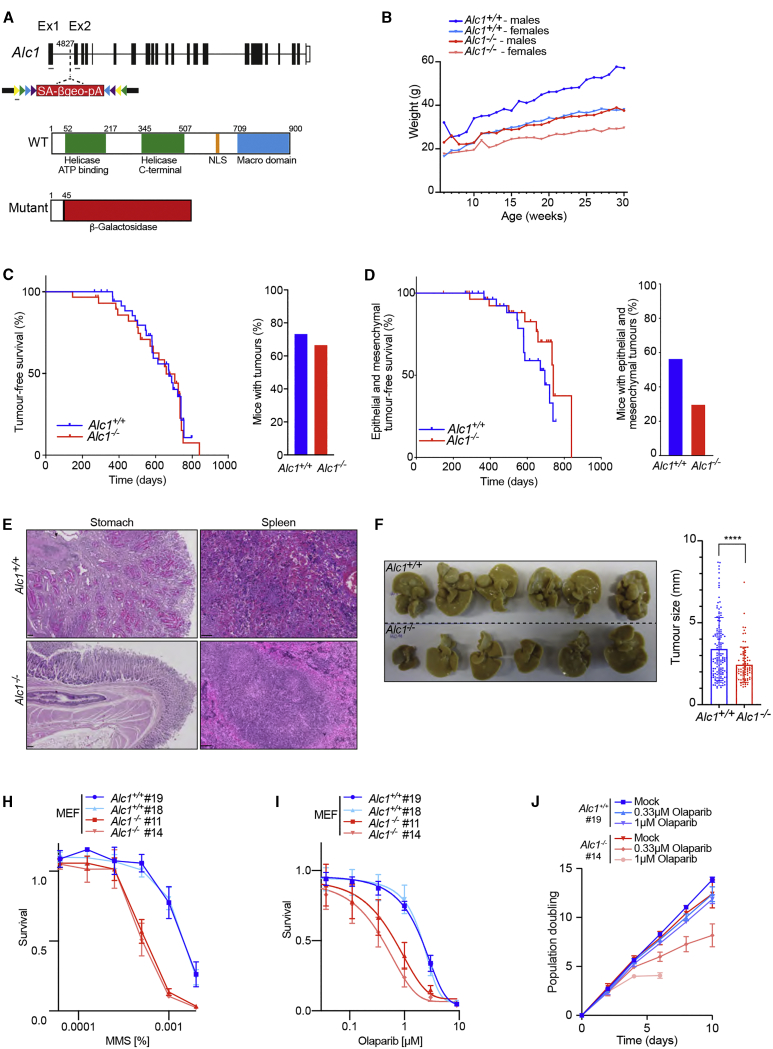
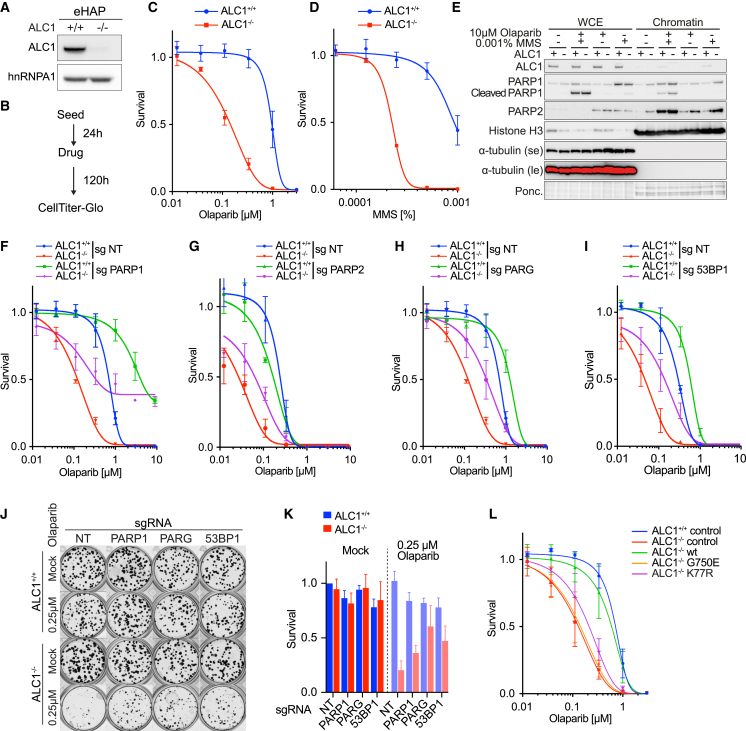
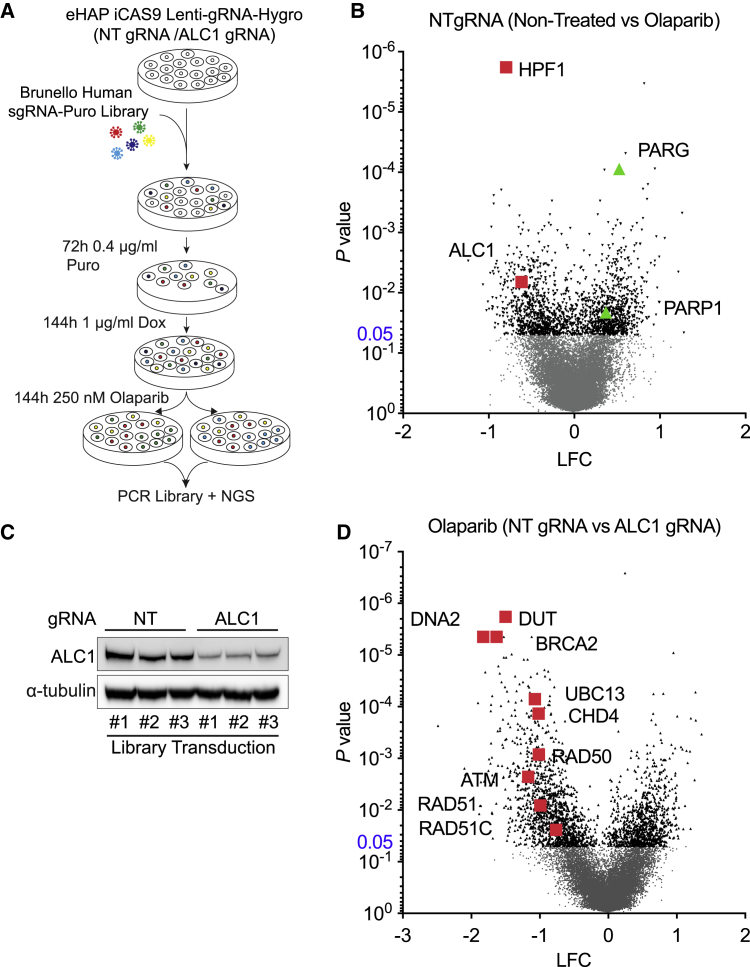
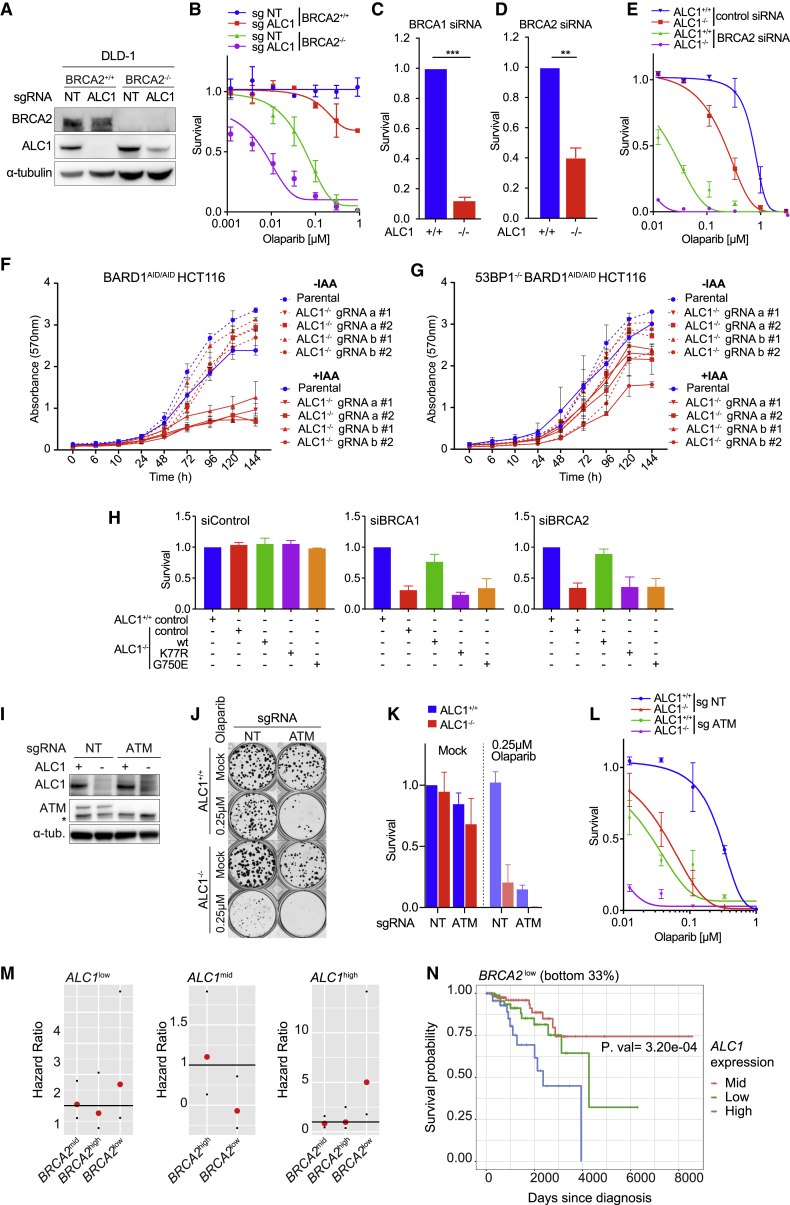
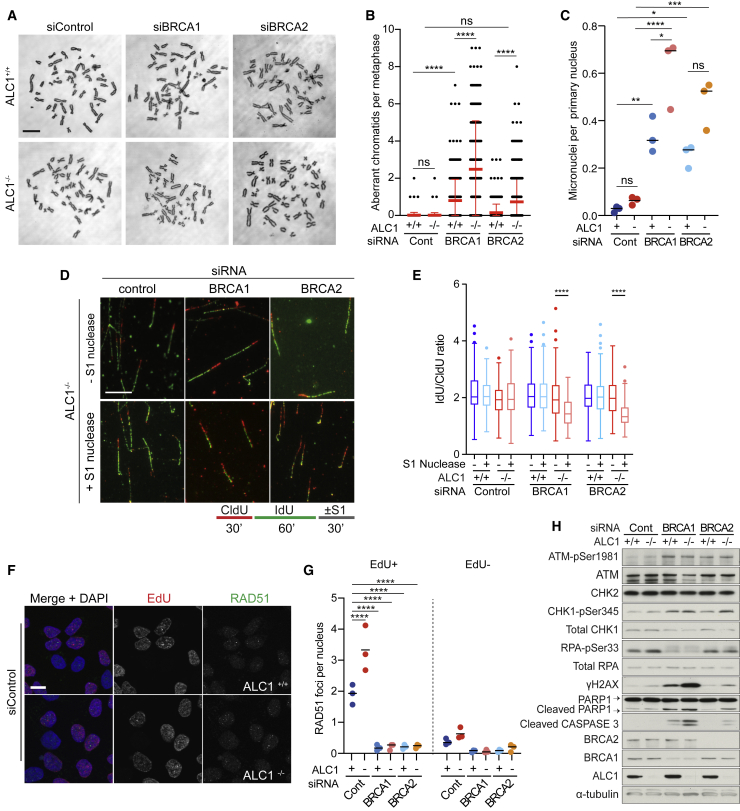
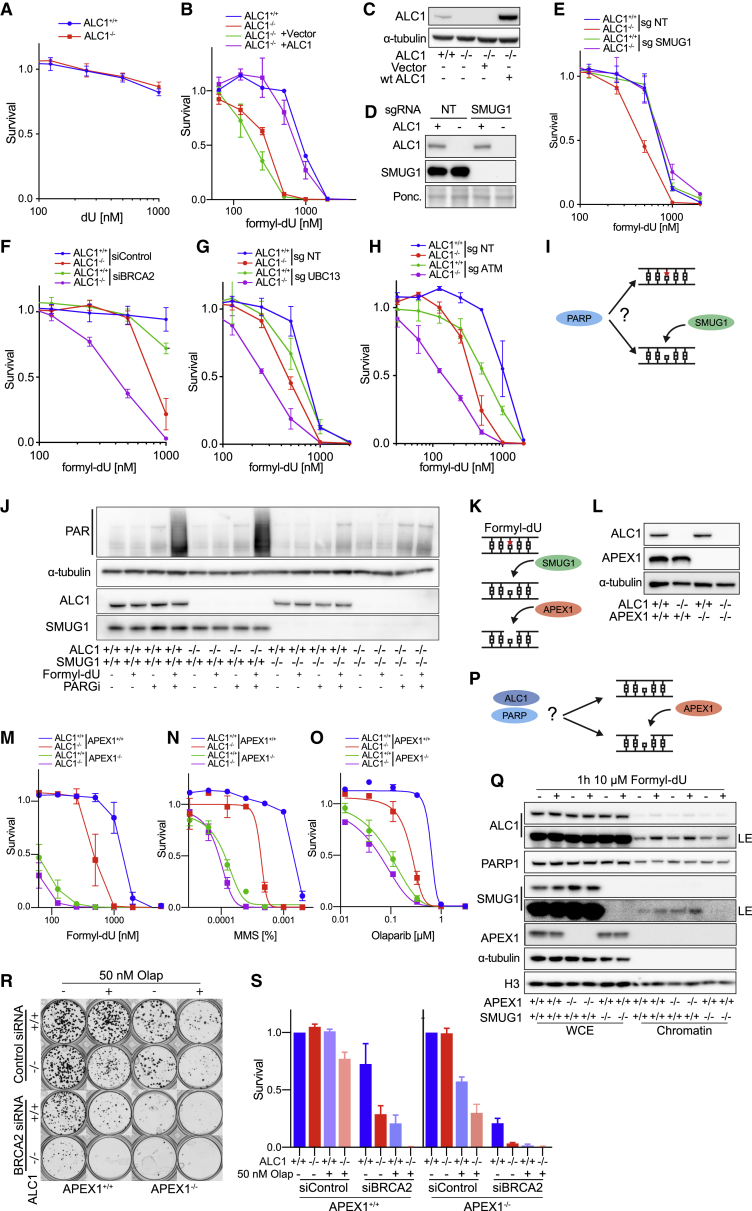
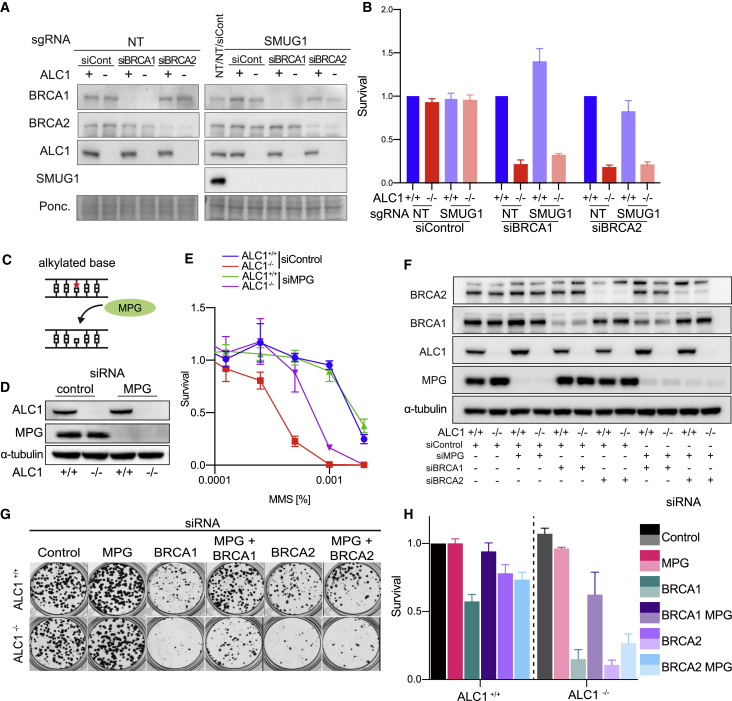
Chromatin is a barrier to efficient DNA repair, as it hinders access and processing of certain DNA lesions. ALC1/CHD1L is a nucleosome-remodeling enzyme that responds to DNA damage, but its precise function in DNA repair remains unknown. Here we report that loss of ALC1 confers sensitivity to PARP inhibitors, methyl-methanesulfonate, and uracil misincorporation, which reflects the need to remodel nucleosomes following base excision by DNA glycosylases but prior to handover to APEX1. Using CRISPR screens, we establish that ALC1 loss is synthetic lethal with homologous recombination deficiency (HRD), which we attribute to chromosome instability caused by unrepaired DNA gaps at replication forks. In the absence of ALC1 or APEX1, incomplete processing of BER intermediates results in post-replicative DNA gaps and a critical dependence on HR for repair. Hence, targeting ALC1 alone or as a PARP inhibitor sensitizer could be employed to augment existing therapeutic strategies for HRD cancers.
Keywords: ALC1; BRCAs; DNA damage repair; DNA gycosylases; PARPs; base excsion repair; chromatin remodeler; homologous recombination defieciency; poly(ADP)-ribosylation; synthetic lethality.
Copyright © 2020 The Author(s). Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration Of Interests G.H. and S.J.B are inventors on a patent derived from this work. S.J.B. is also scientific co-founder and VP Science Strategy at Artios Pharma Ltd., Babraham Research Campus, Cambridge, UK. The authors declare no other competing interests.
Figures
References
-
- Bryant H.E., Schultz N., Thomas H.D., Parker K.M., Flower D., Lopez E., Kyle S., Meuth M., Curtin N.J., Helleday T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005;434:913–917. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- FC0010048/MRC_/Medical Research Council/United Kingdom
- 28990/CRUK_/Cancer Research UK/United Kingdom
- 14276/CRUK_/Cancer Research UK/United Kingdom
- 19270/CRUK_/Cancer Research UK/United Kingdom
- C52690/A19270/CRUK_/Cancer Research UK/United Kingdom
- BB/R007195/1/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- FC0010048/CRUK_/Cancer Research UK/United Kingdom
- C35050/A22284/CRUK_/Cancer Research UK/United Kingdom
- 22284/CRUK_/Cancer Research UK/United Kingdom
- 090532/Z/09/Z/WT_/Wellcome Trust/United Kingdom
- 24439/CRUK_/Cancer Research UK/United Kingdom
- FC0010048/WT_/Wellcome Trust/United Kingdom
- WT_/Wellcome Trust/United Kingdom
- 16304/CRUK_/Cancer Research UK/United Kingdom
- 11582/CRUK_/Cancer Research UK/United Kingdom
- 210634/WT_/Wellcome Trust/United Kingdom
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
