

Post-translational modification analysis of Saccharomyces cerevisiae histone methylation enzymes reveals phosphorylation sites of regulatory potential
- PMID: 33334889
- PMCID: PMC7948420
- DOI: 10.1074/jbc.RA120.015995
Post-translational modification analysis of Saccharomyces cerevisiae histone methylation enzymes reveals phosphorylation sites of regulatory potential
Abstract
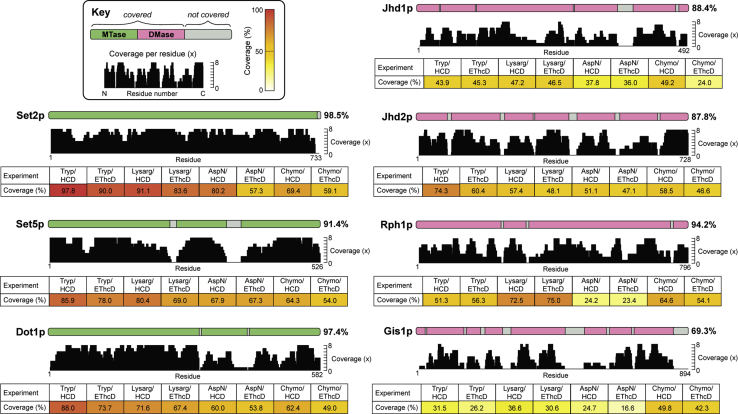
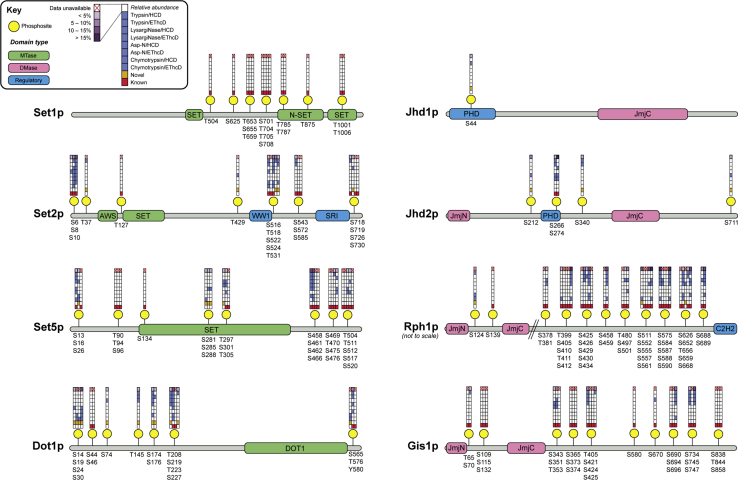
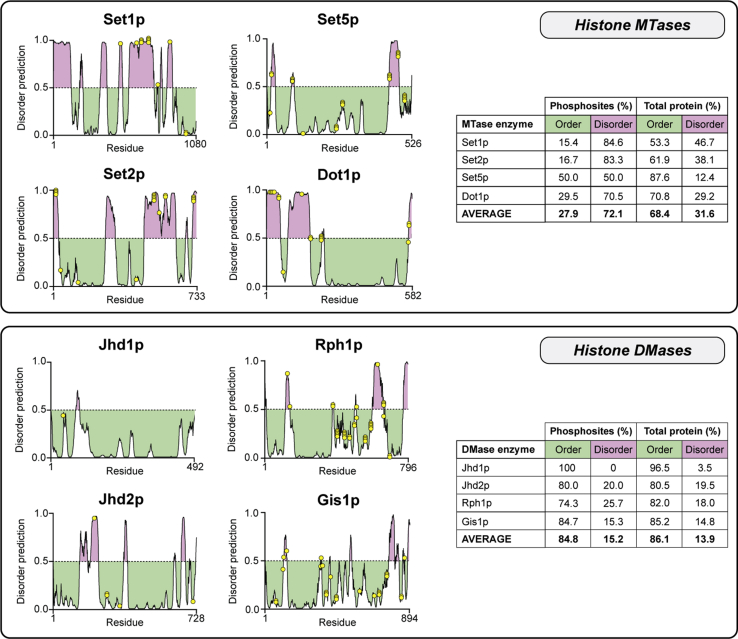
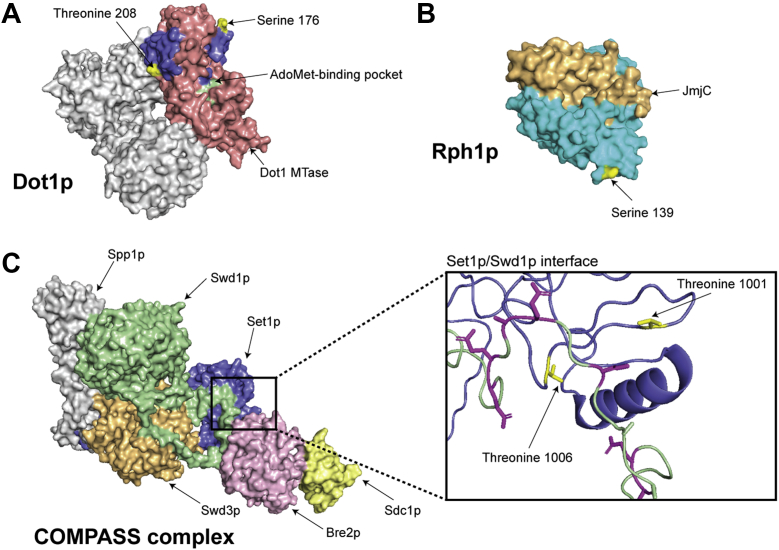
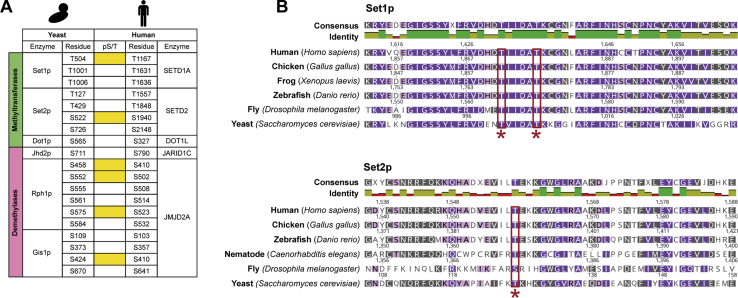
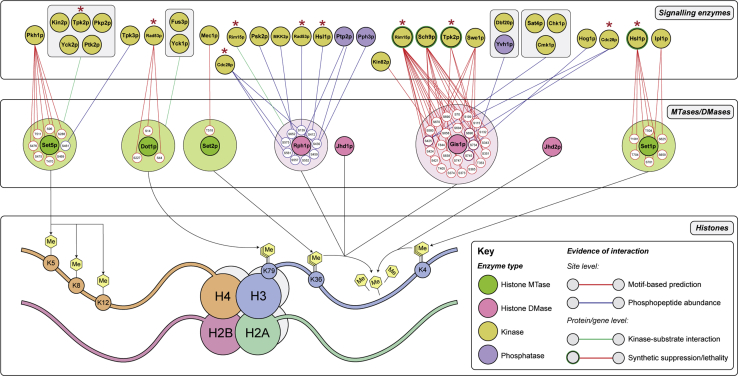
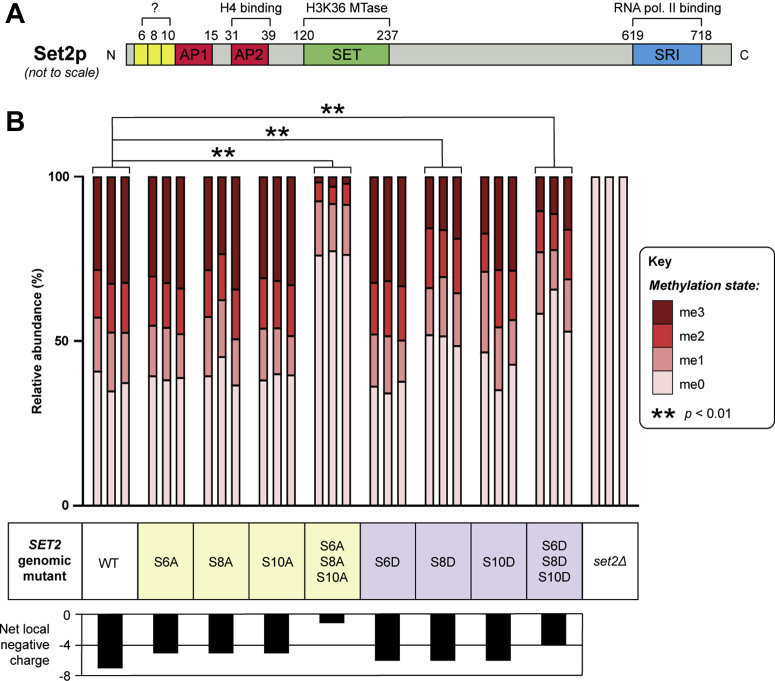
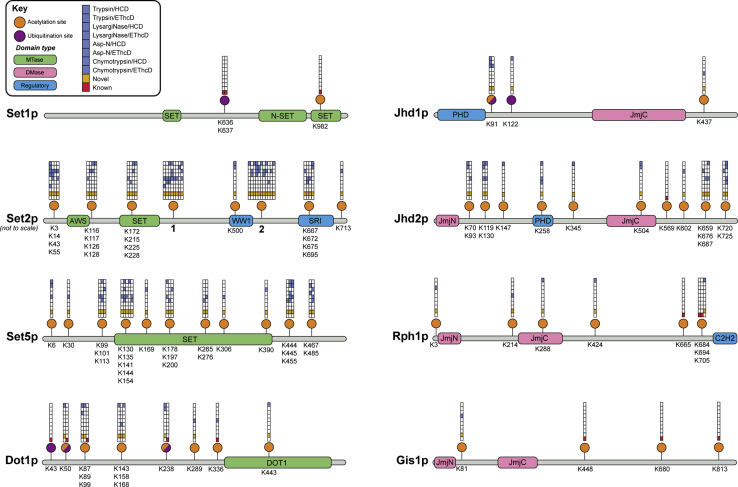
Histone methylation is central to the regulation of eukaryotic transcription. In Saccharomyces cerevisiae, it is controlled by a system of four methyltransferases (Set1p, Set2p, Set5p, and Dot1p) and four demethylases (Jhd1p, Jhd2p, Rph1p, and Gis1p). While the histone targets for these enzymes are well characterized, the connection of the enzymes with the intracellular signaling network and thus their regulation is poorly understood; this also applies to all other eukaryotes. Here we report the detailed characterization of the eight S. cerevisiae enzymes and show that they carry a total of 75 phosphorylation sites, 92 acetylation sites, and two ubiquitination sites. All enzymes are subject to phosphorylation, although demethylases Jhd1p and Jhd2p contained one and five sites respectively, whereas other enzymes carried 14 to 36 sites. Phosphorylation was absent or underrepresented on catalytic and other domains but strongly enriched for regions of disorder on methyltransferases, suggesting a role in the modulation of protein-protein interactions. Through mutagenesis studies, we show that phosphosites within the acidic and disordered N-terminus of Set2p affect H3K36 methylation levels in vivo, illustrating the functional importance of such sites. While most kinases upstream of the yeast histone methylation enzymes remain unknown, we model the possible connections between the cellular signaling network and the histone-based gene regulatory system and propose an integrated regulatory structure. Our results provide a foundation for future, detailed exploration of the role of specific kinases and phosphosites in the regulation of histone methylation.
Keywords: Saccharomyces cerevisiae; chromatin; demethylase; epigenetics; histone methylation; kinase; mass spectrometry; methyltransferase; phosphorylation; post-translational modification.
Copyright © 2021 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Conflict of interest The authors declare that they have no conflicts of interest with the contents of this article.
Figures
References
-
- Martin C., Zhang Y. The diverse functions of histone lysine methylation. Nat. Rev. Mol. Cell Biol. 2005;6:838–849. - PubMed
-
- Margueron R., Trojer P., Reinberg D. The key to development: interpreting the histone code? Curr. Opin. Genet. Dev. 2005;15:163–176. - PubMed
-
- Shi Y., Whetstine J.R. Dynamic regulation of histone lysine methylation by demethylases. Mol. Cell. 2007;25:1–14. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
