

Sequence diversity analyses of an improved rhesus macaque genome enhance its biomedical utility
- PMID: 33335035
- PMCID: PMC7818670
- DOI: 10.1126/science.abc6617
Sequence diversity analyses of an improved rhesus macaque genome enhance its biomedical utility
Abstract
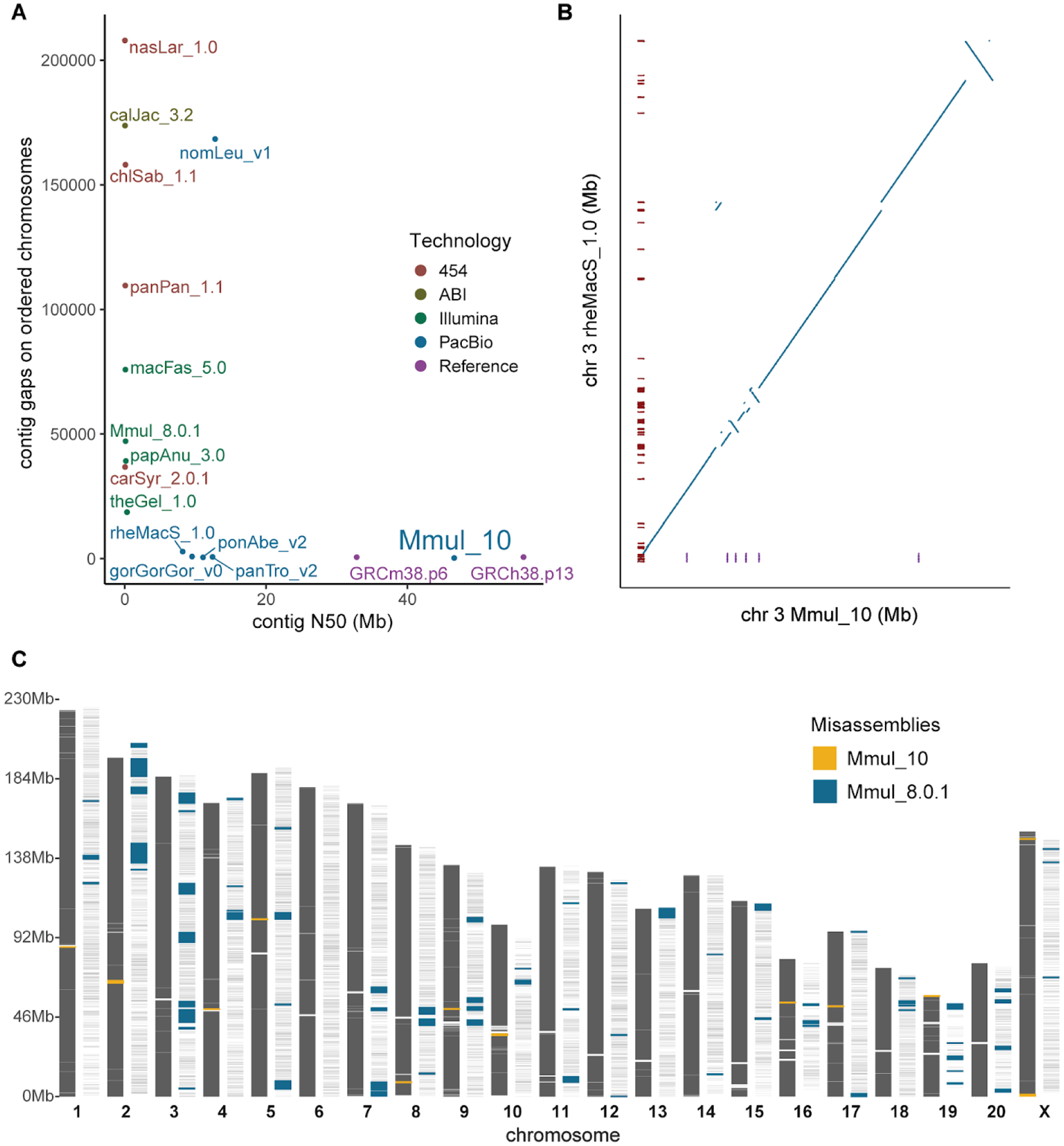
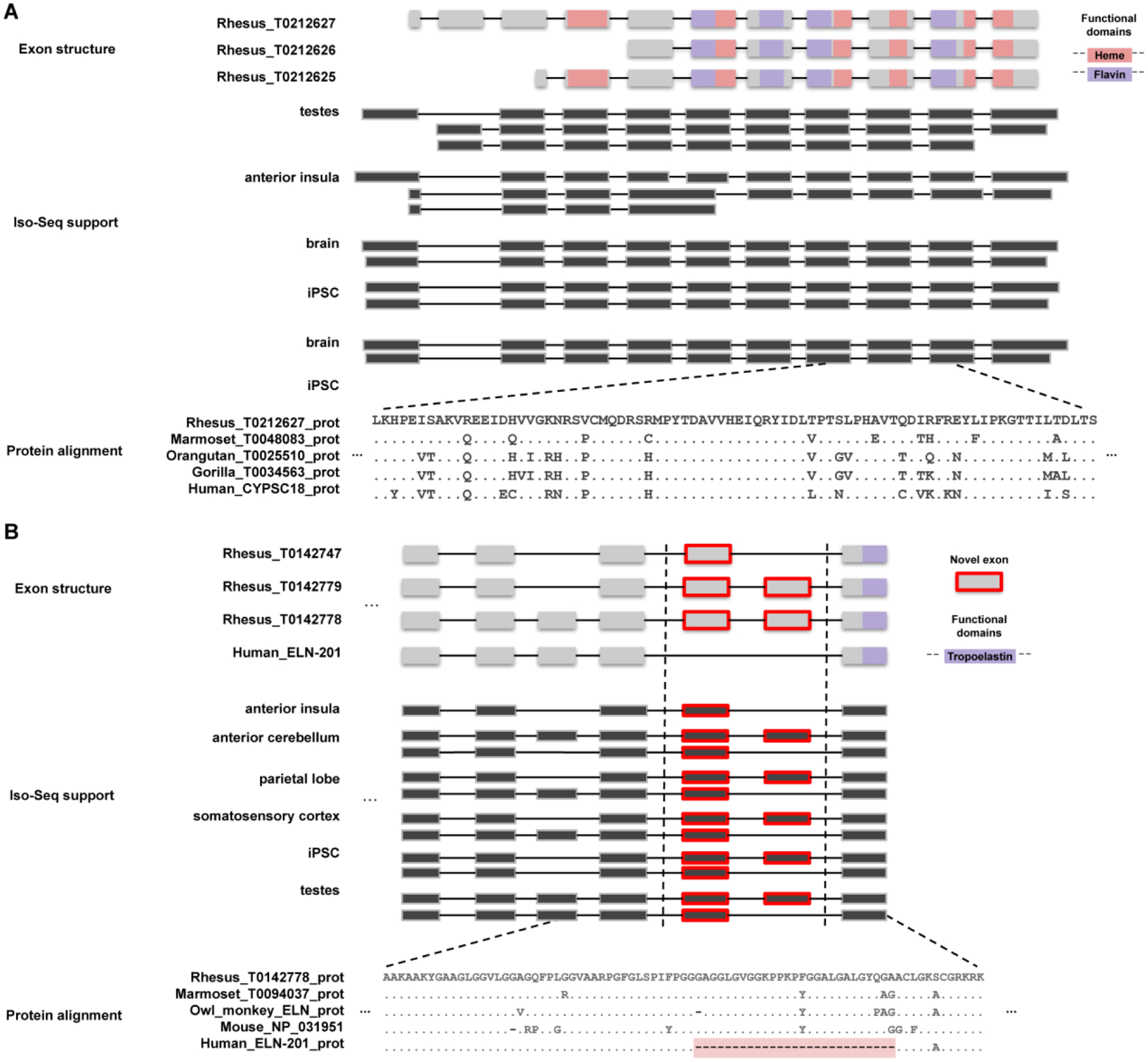
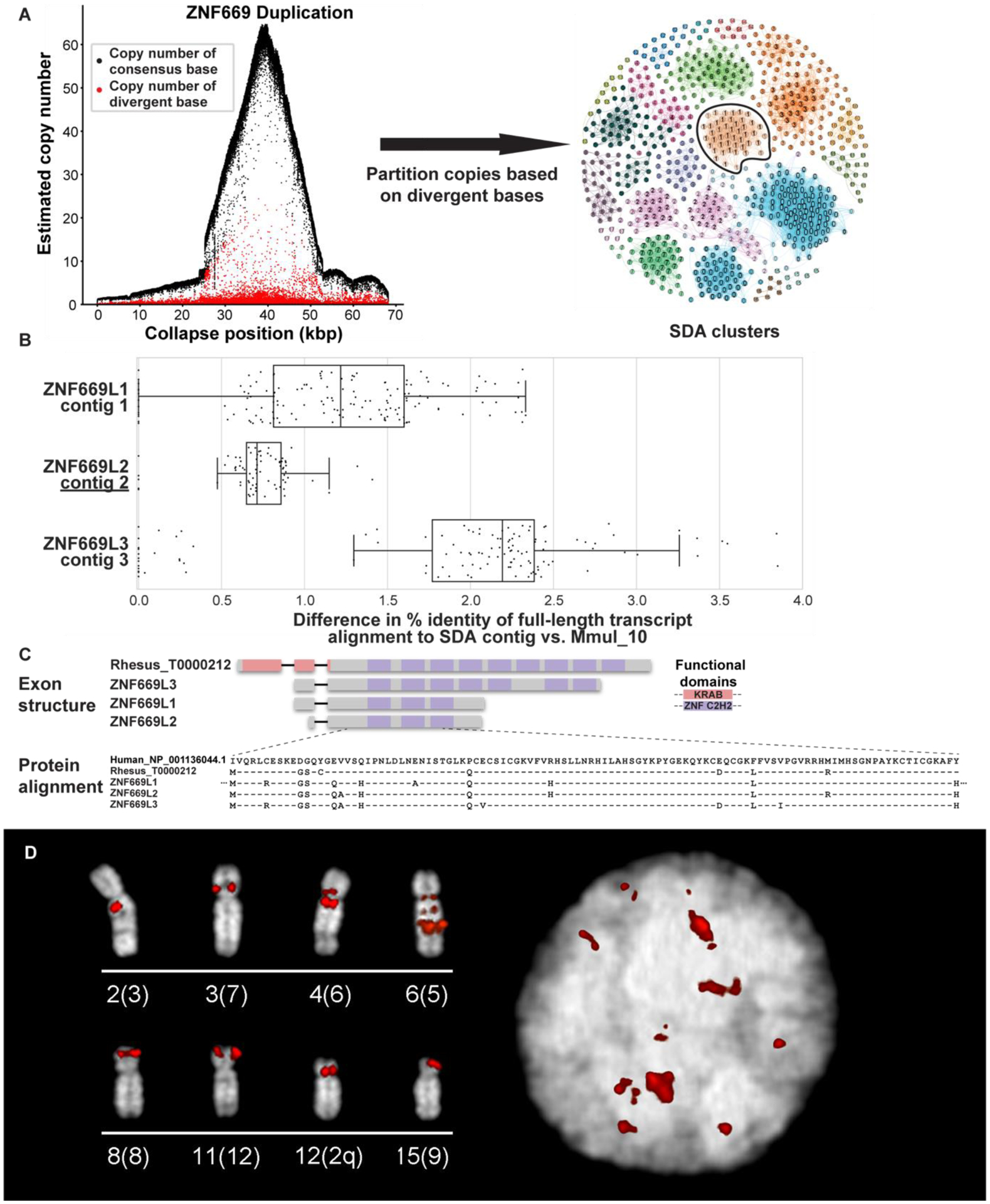
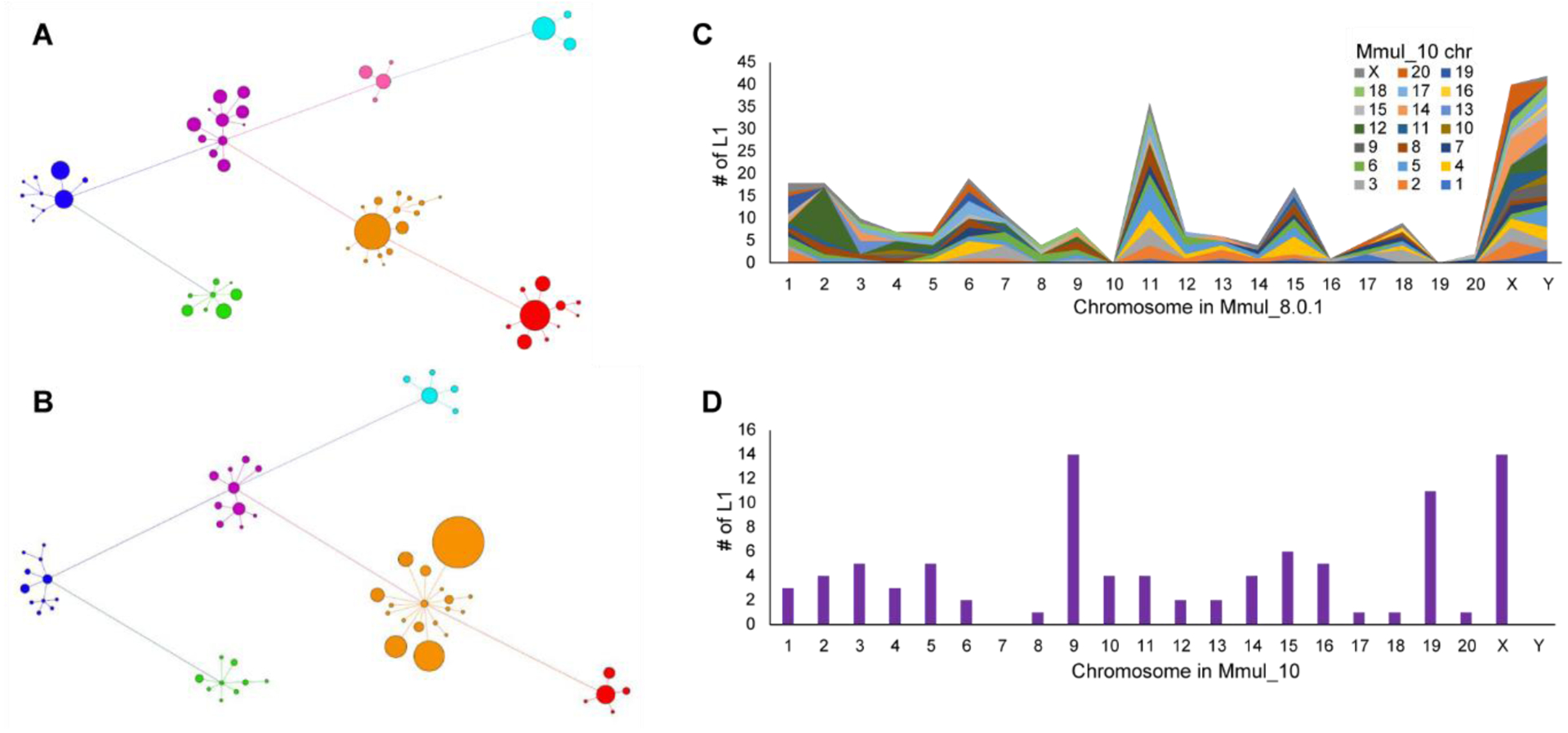
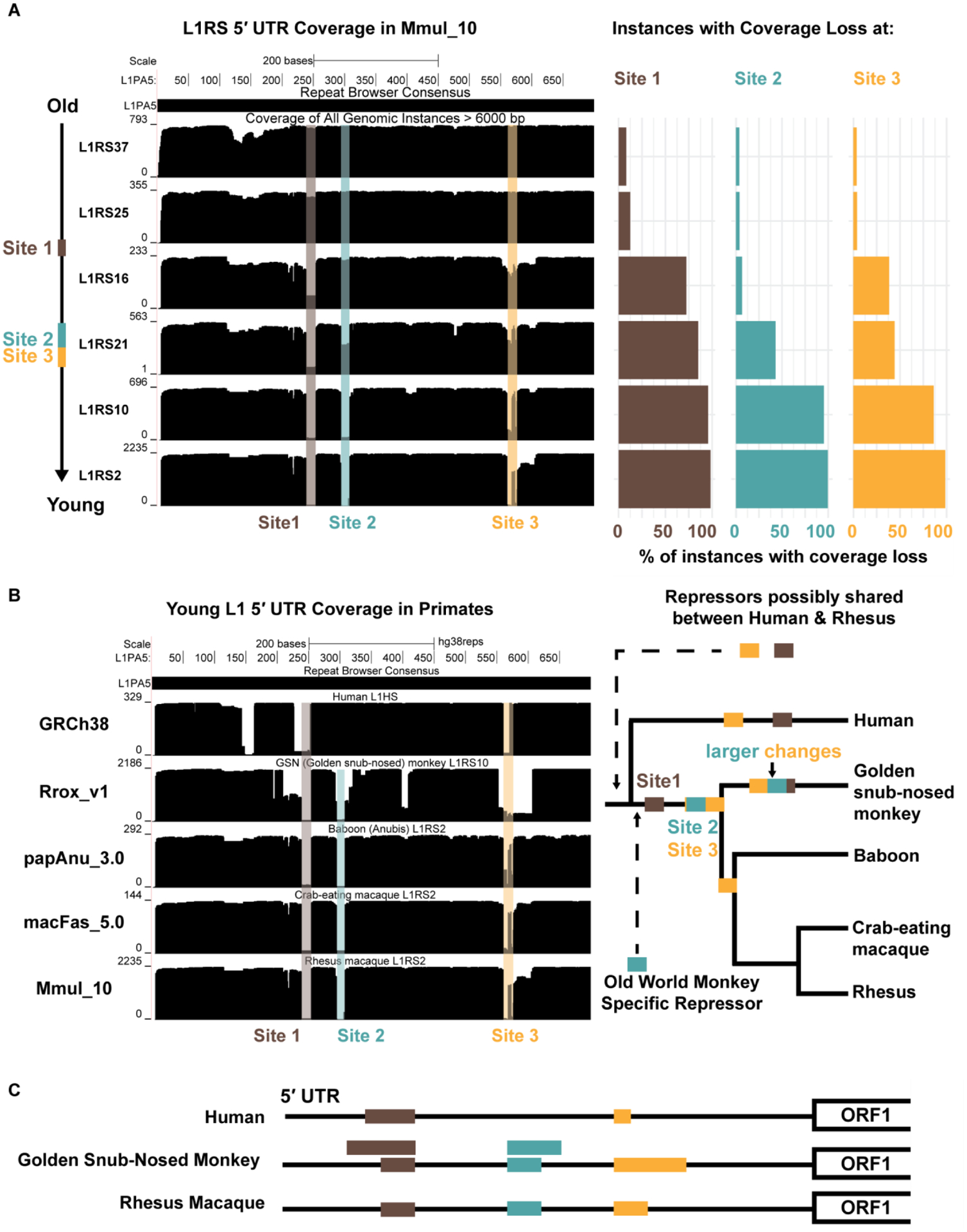
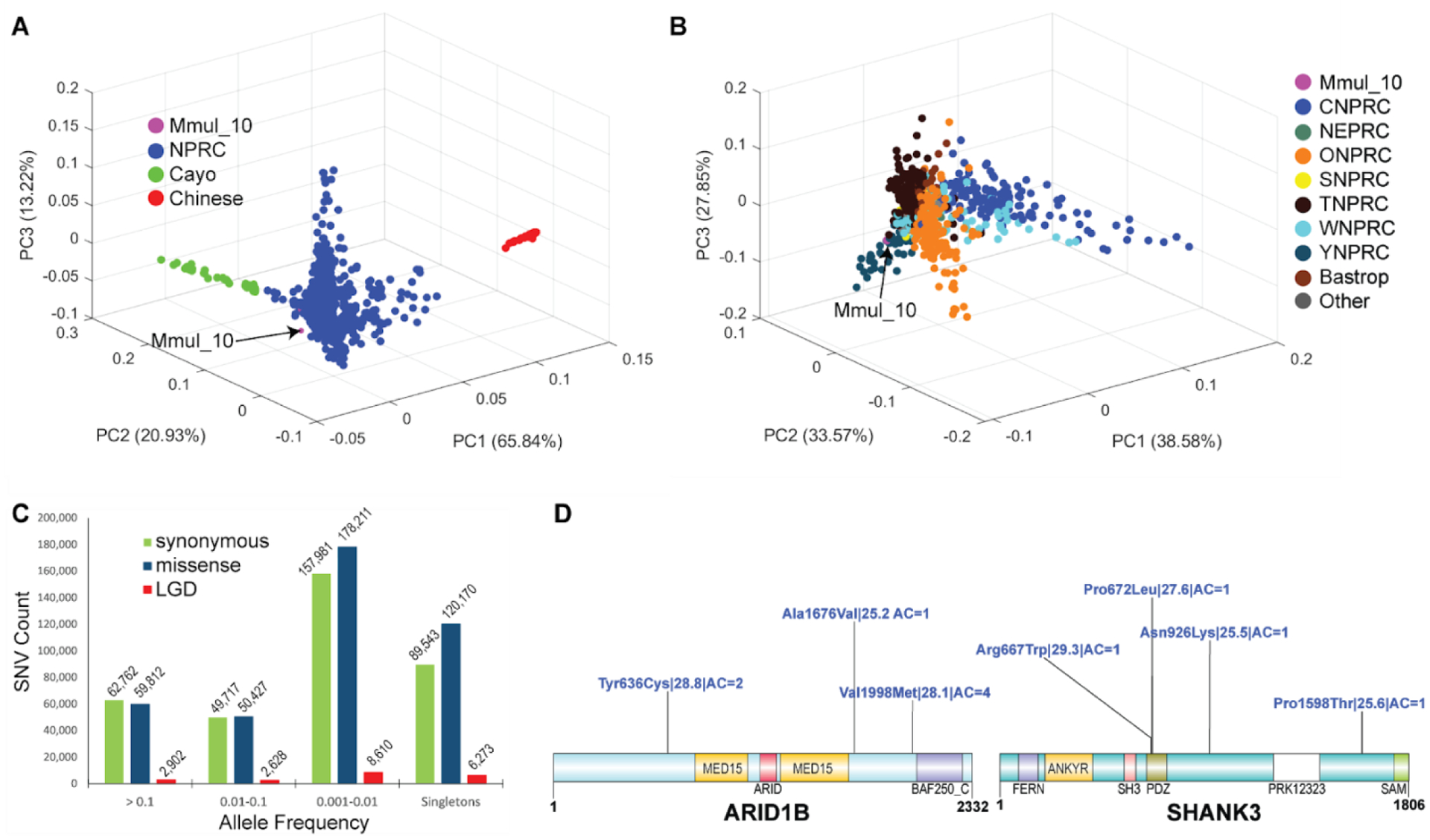
The rhesus macaque (Macaca mulatta) is the most widely studied nonhuman primate (NHP) in biomedical research. We present an updated reference genome assembly (Mmul_10, contig N50 = 46 Mbp) that increases the sequence contiguity 120-fold and annotate it using 6.5 million full-length transcripts, thus improving our understanding of gene content, isoform diversity, and repeat organization. With the improved assembly of segmental duplications, we discovered new lineage-specific genes and expanded gene families that are potentially informative in studies of evolution and disease susceptibility. Whole-genome sequencing (WGS) data from 853 rhesus macaques identified 85.7 million single-nucleotide variants (SNVs) and 10.5 million indel variants, including potentially damaging variants in genes associated with human autism and developmental delay, providing a framework for developing noninvasive NHP models of human disease.
Copyright © 2020 The Authors, some rights reserved; exclusive licensee American Association for the Advancement of Science. No claim to original U.S. Government Works.
Conflict of interest statement
Figures
References
Publication types
MeSH terms
Grants and funding
- U01 HL137183/HL/NHLBI NIH HHS/United States
- P51 OD011107/OD/NIH HHS/United States
- P51 OD011104/OD/NIH HHS/United States
- T32 HG008345/HG/NHGRI NIH HHS/United States
- R01 HG002939/HG/NHGRI NIH HHS/United States
- R01 GM059290/GM/NIGMS NIH HHS/United States
- P50 MH100031/MH/NIMH NIH HHS/United States
- P51 OD011092/OD/NIH HHS/United States
- U01 HG010961/HG/NHGRI NIH HHS/United States
- P51 OD011132/OD/NIH HHS/United States
- U41 HG010972/HG/NHGRI NIH HHS/United States
- U42 OD024282/OD/NIH HHS/United States
- HHMI/Howard Hughes Medical Institute/United States
- R24 OD010962/OD/NIH HHS/United States
- T32 HG000035/HG/NHGRI NIH HHS/United States
- U41 HG007234/HG/NHGRI NIH HHS/United States
- R01 HG010329/HG/NHGRI NIH HHS/United States
- U24 HG009081/HG/NHGRI NIH HHS/United States
- R24 OD021324/OD/NIH HHS/United States
- R01 MH081884/MH/NIMH NIH HHS/United States
- R01 HG010485/HG/NHGRI NIH HHS/United States
- U42 OD010568/OD/NIH HHS/United States
- U54 HG007990/HG/NHGRI NIH HHS/United States
- R24 OD011173/OD/NIH HHS/United States
- R01 HG002385/HG/NHGRI NIH HHS/United States
- P51 OD011106/OD/NIH HHS/United States
- R01 MH046729/MH/NIMH NIH HHS/United States
- U24 HG009084/HG/NHGRI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
