

Allosteric inhibition explained through conformational ensembles sampling distinct "mixed" states
- PMID: 33335680
- PMCID: PMC7720024
- DOI: 10.1016/j.csbj.2020.10.026
Allosteric inhibition explained through conformational ensembles sampling distinct "mixed" states
Abstract
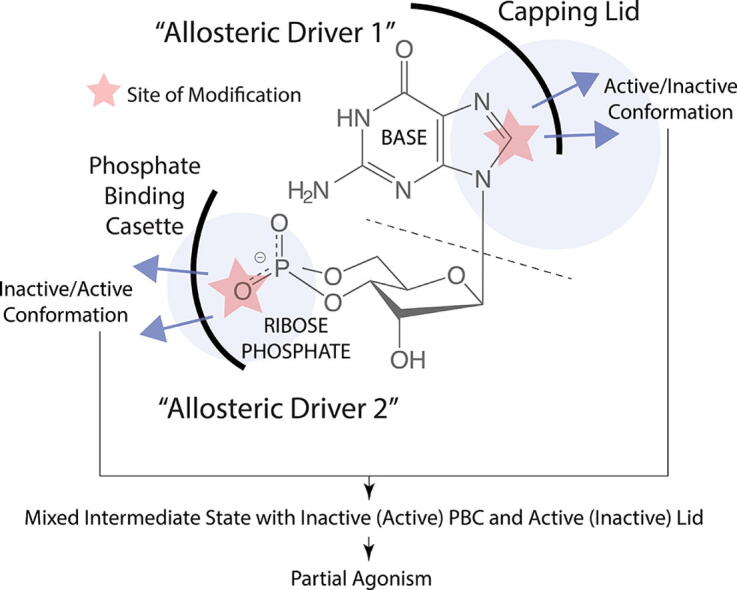
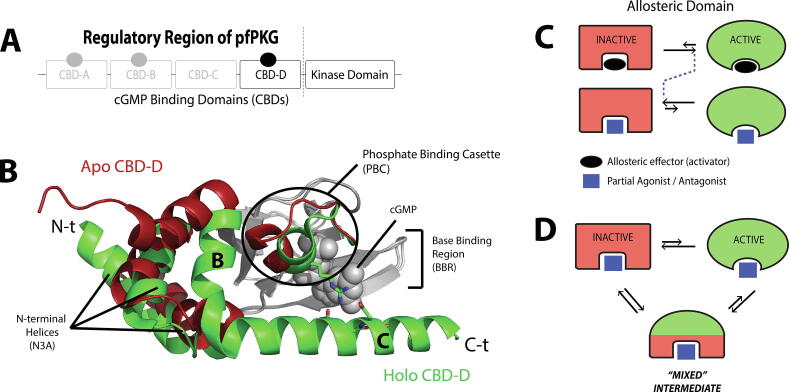
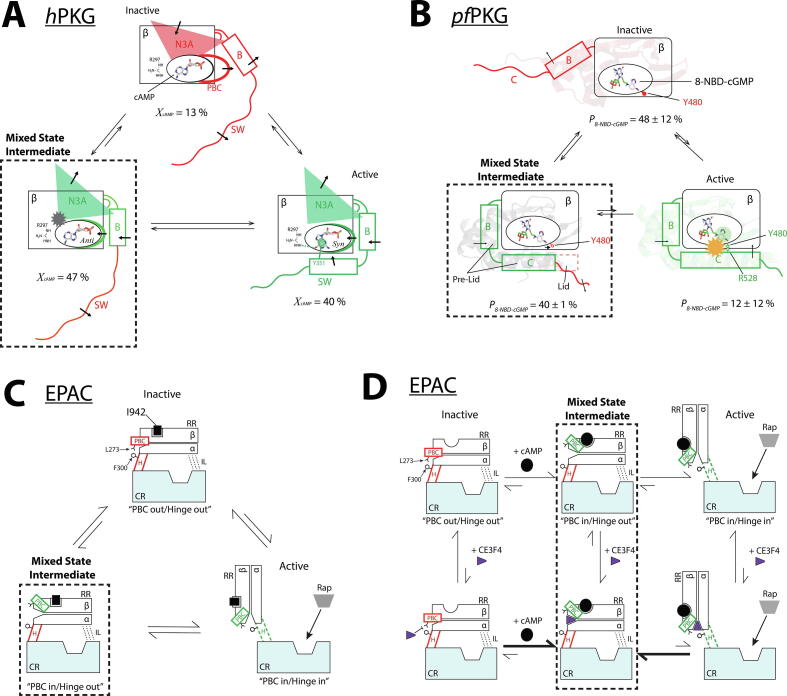
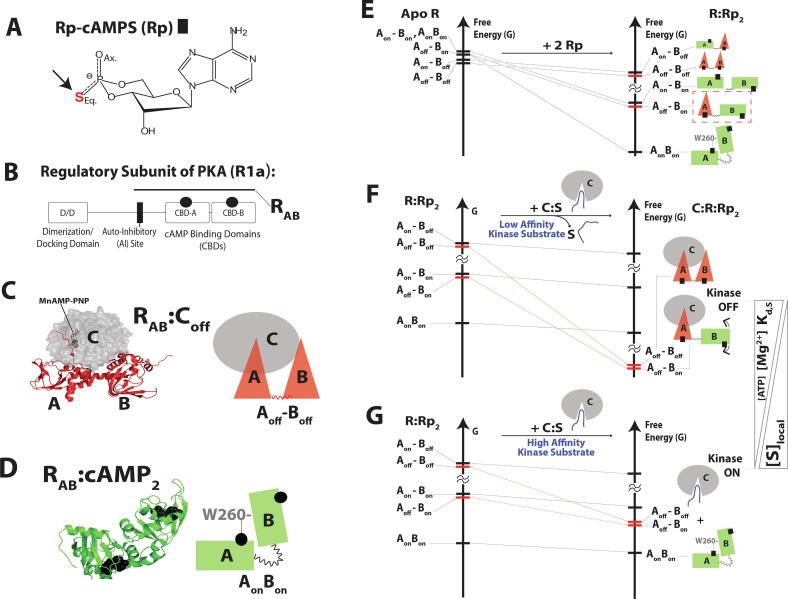
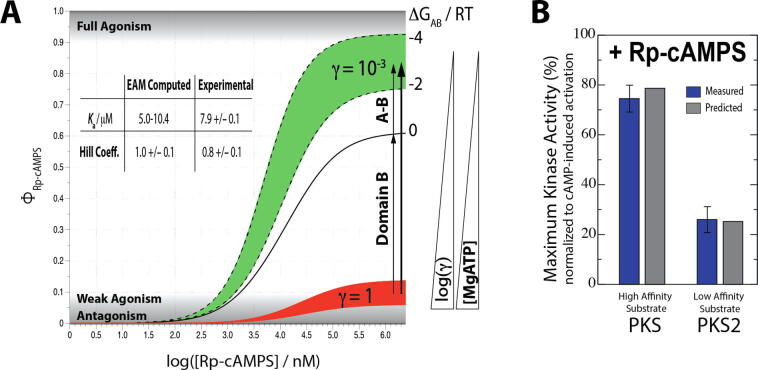
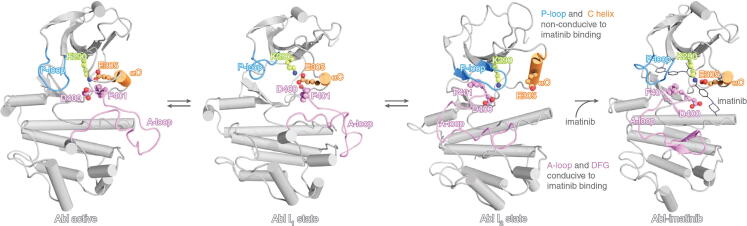
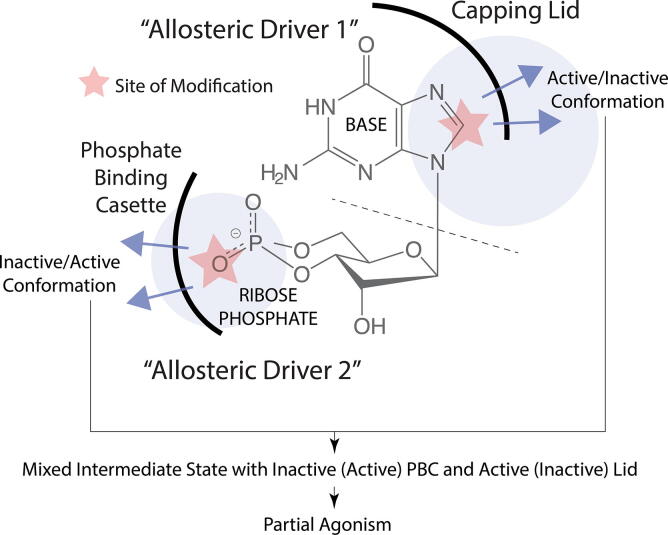
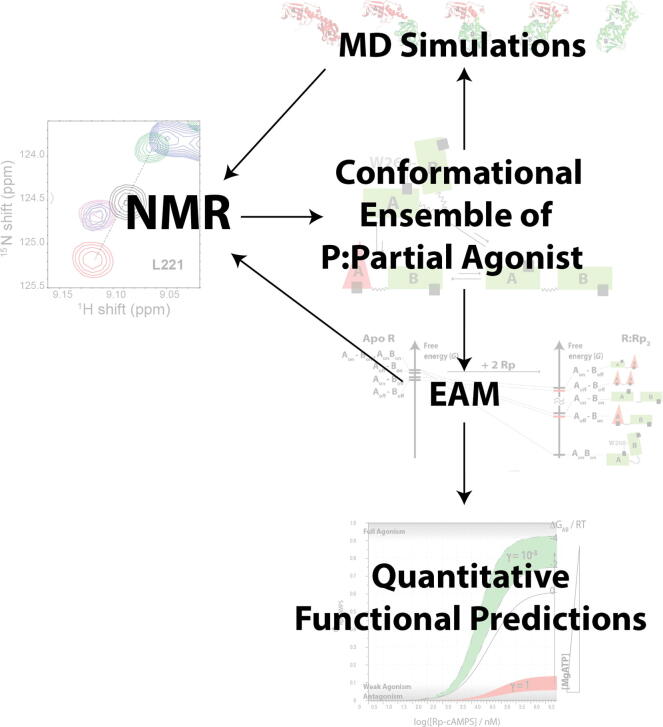
Allosteric modulation provides an effective avenue for selective and potent enzyme inhibition. Here, we summarize and critically discuss recent advances on the mechanisms of allosteric partial agonists for three representative signalling enzymes activated by cyclic nucleotides: the cAMP-dependent protein kinase (PKA), the cGMP-dependent protein kinase (PKG), and the exchange protein activated by cAMP (EPAC). The comparative analysis of partial agonism in PKA, PKG and EPAC reveals a common emerging theme, i.e. the sampling of distinct "mixed" conformational states, either within a single domain or between distinct domains. Here, we show how such "mixed" states play a crucial role in explaining the observed functional response, i.e. partial agonism and allosteric pluripotency, as well as in maximizing inhibition while minimizing potency losses. In addition, by combining Nuclear Magnetic Resonance (NMR), Molecular Dynamics (MD) simulations and Ensemble Allosteric Modeling (EAM), we also show how to map the free-energy landscape of conformational ensembles containing "mixed" states. By discussing selected case studies, we illustrate how MD simulations and EAM complement NMR to quantitatively relate protein dynamics to function. The resulting NMR- and MD-based EAMs are anticipated to inform not only the design of new generations of highly selective allosteric inhibitors, but also the choice of multidrug combinations.
Keywords: Agonism; Allosteric pluripotency; Allostery; Antagonism; EPAC; PKA; PKG; cAMP; cGMP.
© 2020 The Authors.
Conflict of interest statement
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Figures
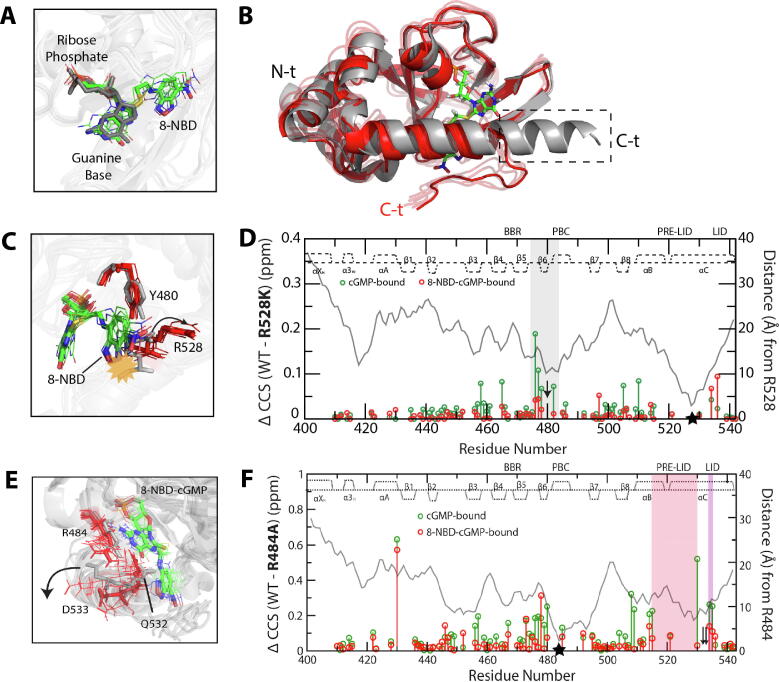
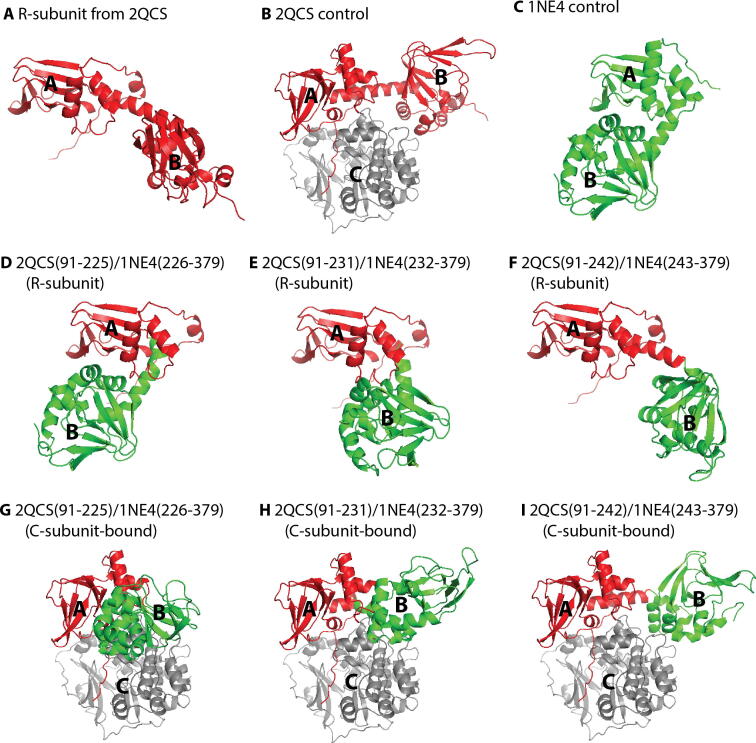
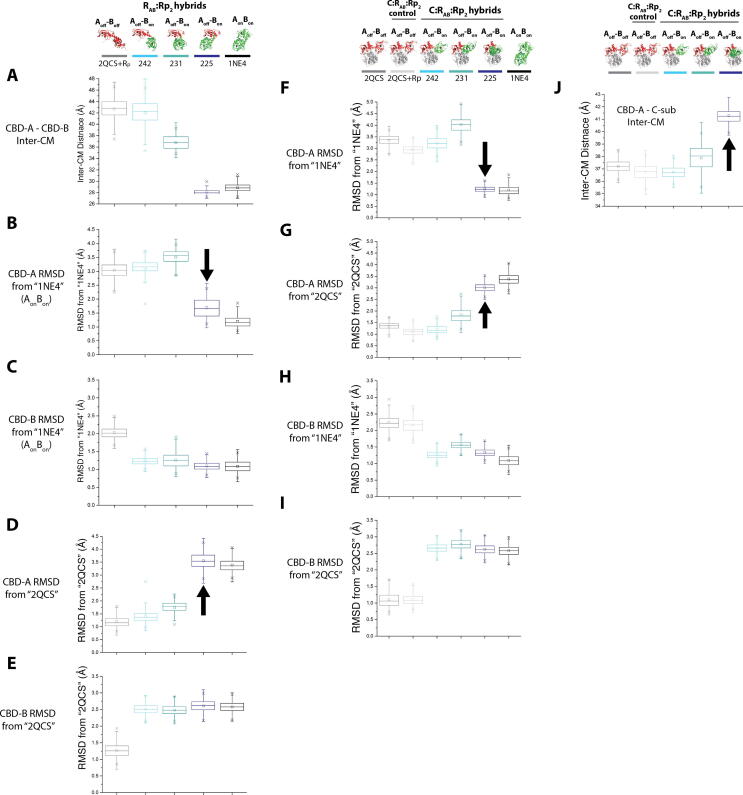
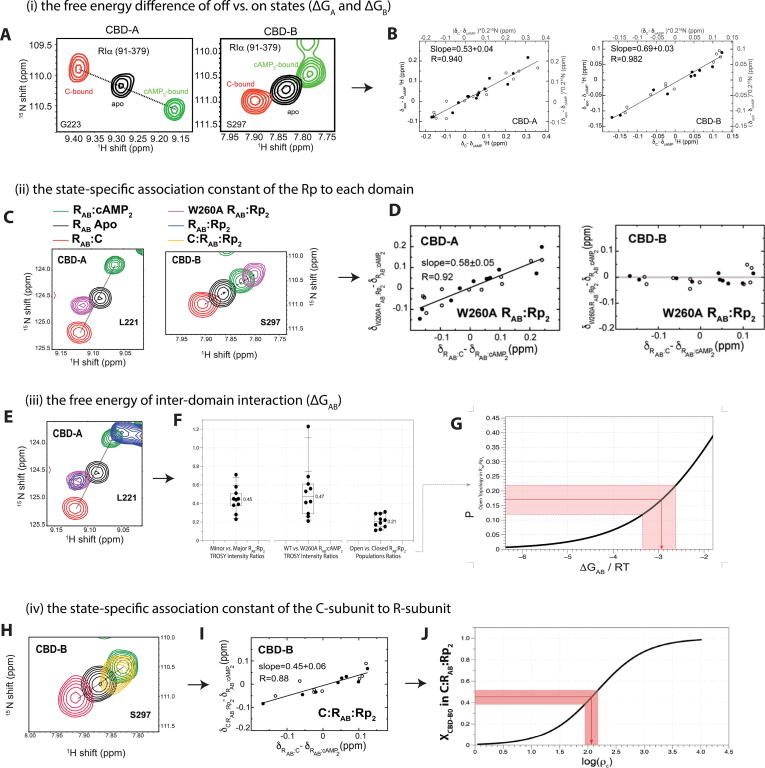
References
Publication types
LinkOut - more resources
Full Text Sources