

scTenifoldNet: A Machine Learning Workflow for Constructing and Comparing Transcriptome-wide Gene Regulatory Networks from Single-Cell Data
- PMID: 33336197
- PMCID: PMC7733883
- DOI: 10.1016/j.patter.2020.100139
scTenifoldNet: A Machine Learning Workflow for Constructing and Comparing Transcriptome-wide Gene Regulatory Networks from Single-Cell Data
Abstract
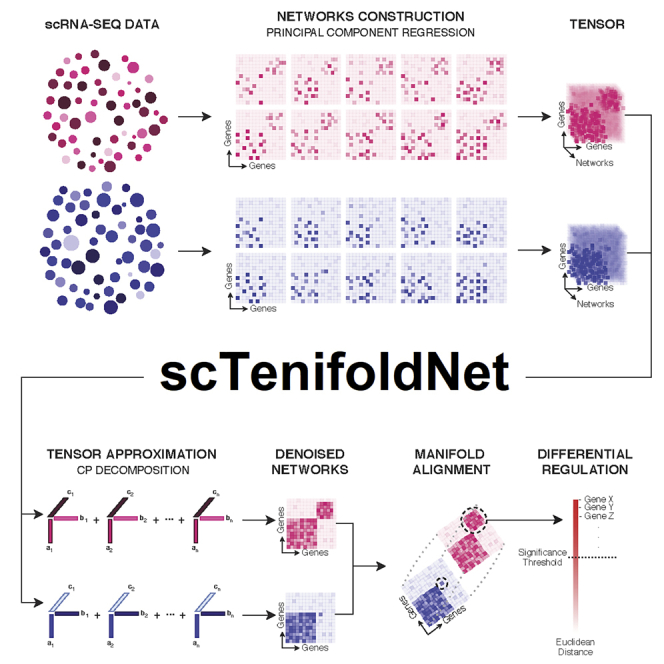
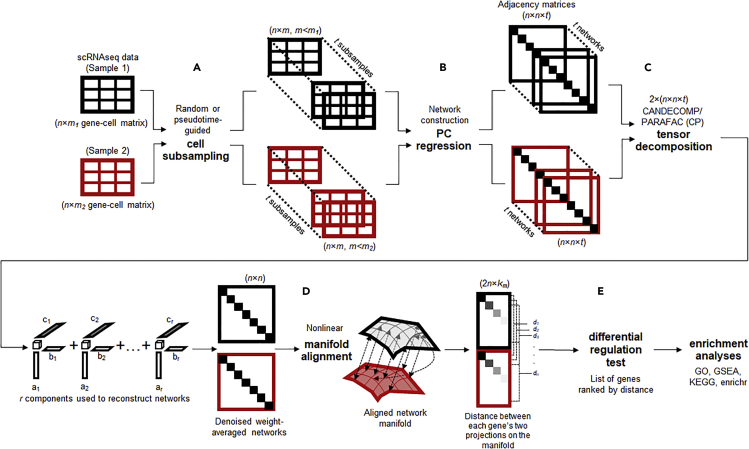
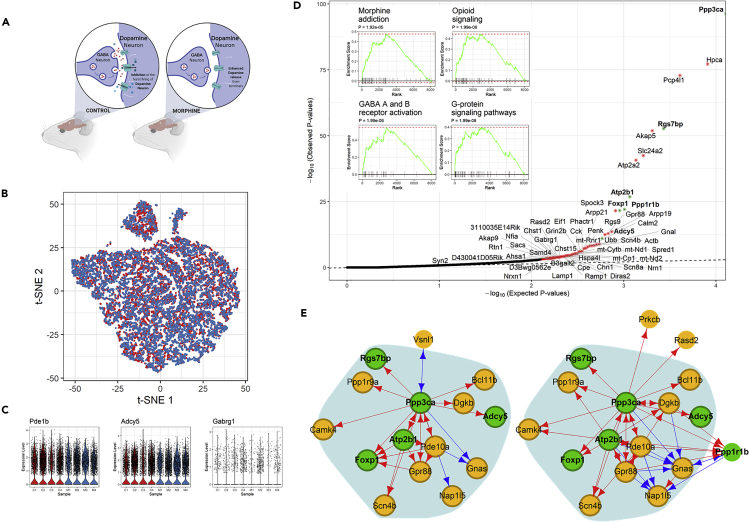
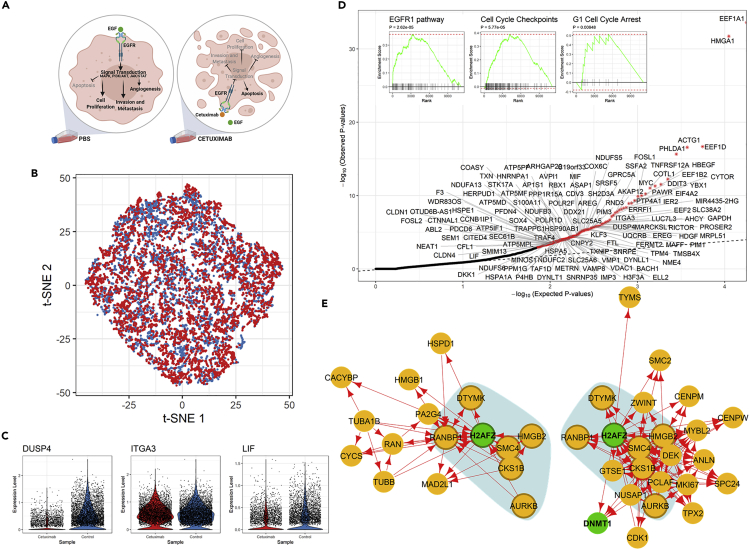
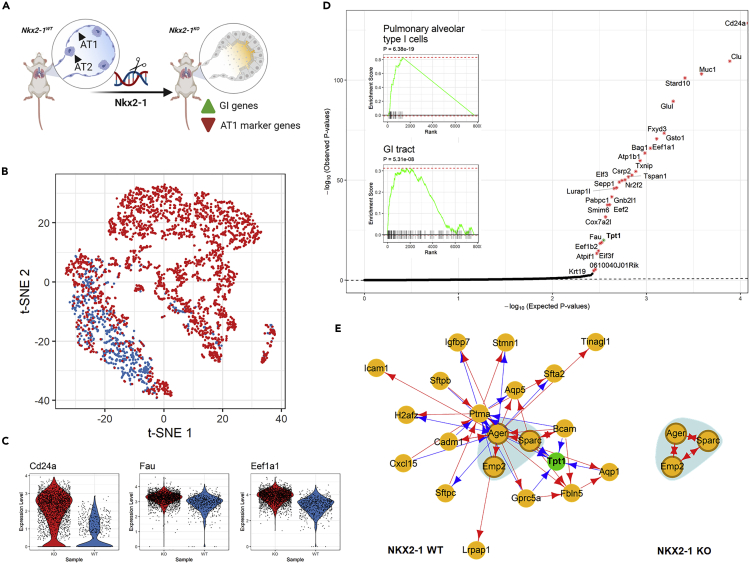
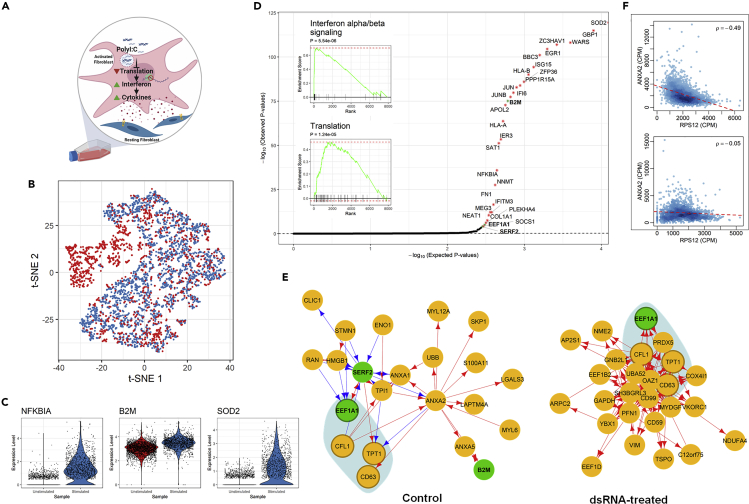
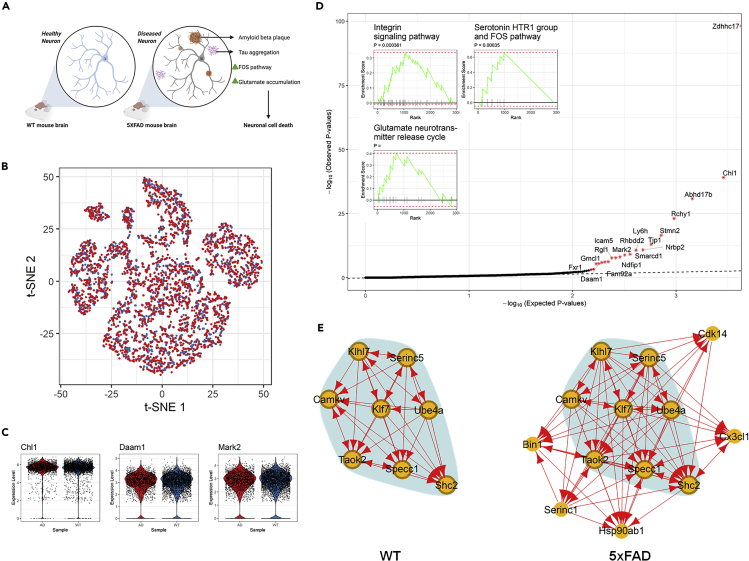
We present scTenifoldNet-a machine learning workflow built upon principal-component regression, low-rank tensor approximation, and manifold alignment-for constructing and comparing single-cell gene regulatory networks (scGRNs) using data from single-cell RNA sequencing. scTenifoldNet reveals regulatory changes in gene expression between samples by comparing the constructed scGRNs. With real data, scTenifoldNet identifies specific gene expression programs associated with different biological processes, providing critical insights into the underlying mechanism of regulatory networks governing cellular transcriptional activities.
Keywords: gene regulatory network; machine learning; manifold alignment; principal-component regression; scRNA-seq; scTenifoldNet; single-cell RNA sequencing; tensor decomposition.
© 2020 The Authors.
Conflict of interest statement
The authors declare no competing interests.
Figures

References
-
- Friedman N., Linial M., Nachman I., Pe'er D. Using Bayesian networks to analyze expression data. J. Comput. Biol. 2000;7:601–620. - PubMed
LinkOut - more resources
Full Text Sources
