

Pathophysiology and clinical consequences of arterial blood gases and pH after cardiac arrest
- PMID: 33336311
- PMCID: PMC7746422
- DOI: 10.1186/s40635-020-00307-1
Pathophysiology and clinical consequences of arterial blood gases and pH after cardiac arrest
Abstract
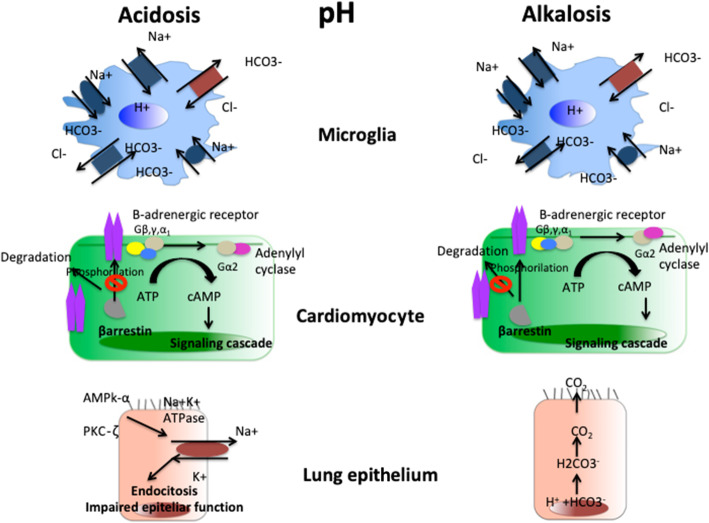
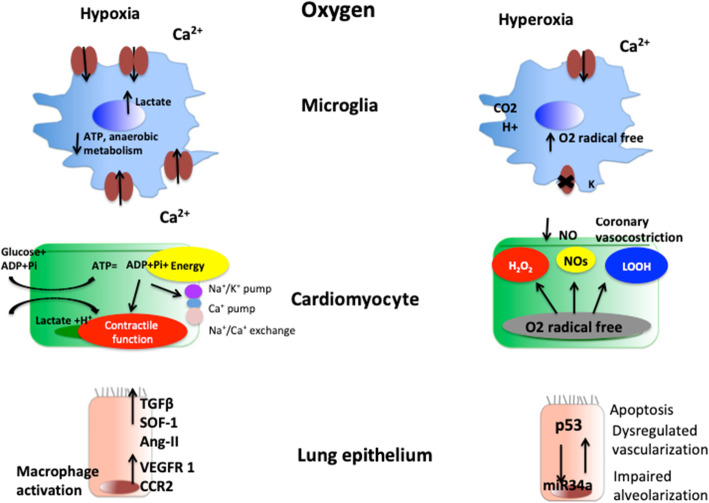
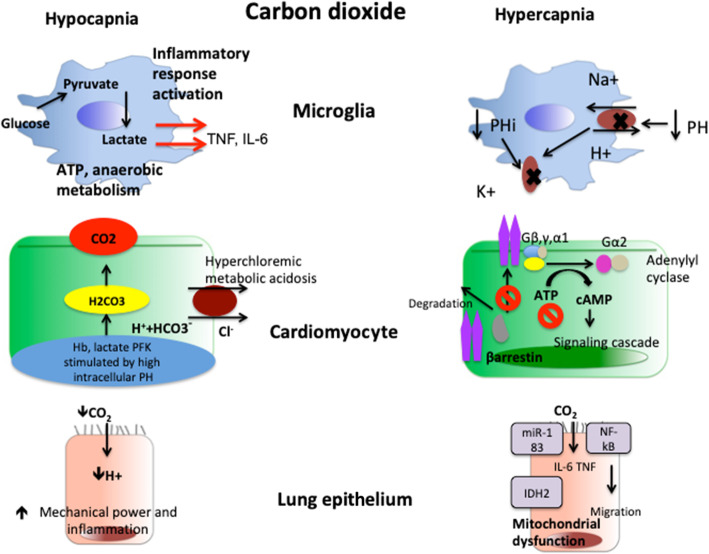
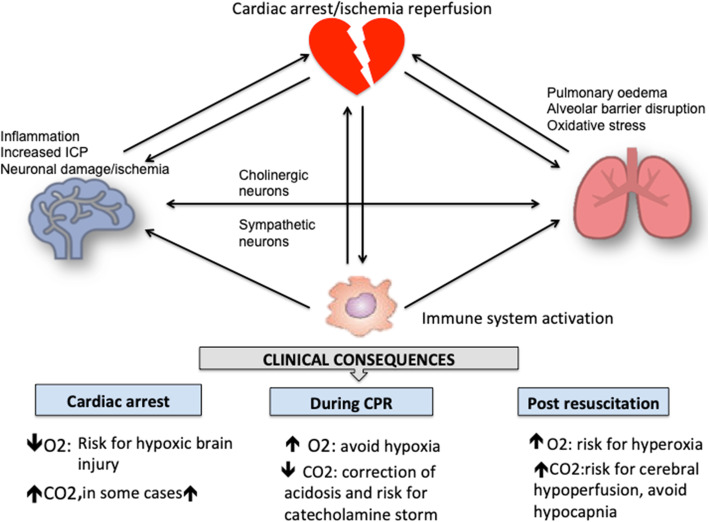
Post cardiac arrest syndrome is associated with high morbidity and mortality, which is related not only to a poor neurological outcome but also to respiratory and cardiovascular dysfunctions. The control of gas exchange, and in particular oxygenation and carbon dioxide levels, is fundamental in mechanically ventilated patients after resuscitation, as arterial blood gases derangement might have important effects on the cerebral blood flow and systemic physiology.In particular, the pathophysiological role of carbon dioxide (CO2) levels is strongly underestimated, as its alterations quickly affect also the changes of intracellular pH, and consequently influence metabolic energy and oxygen demand. Hypo/hypercapnia, as well as mechanical ventilation during and after resuscitation, can affect CO2 levels and trigger a dangerous pathophysiological vicious circle related to the relationship between pH, cellular demand, and catecholamine levels. The developing hypocapnia can nullify the beneficial effects of the hypothermia. The aim of this review was to describe the pathophysiology and clinical consequences of arterial blood gases and pH after cardiac arrest.According to our findings, the optimal ventilator strategies in post cardiac arrest patients are not fully understood, and oxygen and carbon dioxide targets should take in consideration a complex pattern of pathophysiological factors. Further studies are warranted to define the optimal settings of mechanical ventilation in patients after cardiac arrest.
Keywords: Cardiac arrest; Catecholamine; Gas exchanges; Intracellular acidosis; Ventilator targets.
Conflict of interest statement
None
Figures

References
-
- Phypers B, Pierce JT. Lactate physiology in health and disease. Contin Educ Anaesth Crit Care Pain. 2006;6:128–132. doi: 10.1093/bjaceaccp/mkl018. - DOI
Publication types
LinkOut - more resources
Full Text Sources
