

A Surge of DNA Damage Links Transcriptional Reprogramming and Hematopoietic Deficit in Fanconi Anemia
- PMID: 33338401
- PMCID: PMC8600940
- DOI: 10.1016/j.molcel.2020.11.040
A Surge of DNA Damage Links Transcriptional Reprogramming and Hematopoietic Deficit in Fanconi Anemia
Abstract
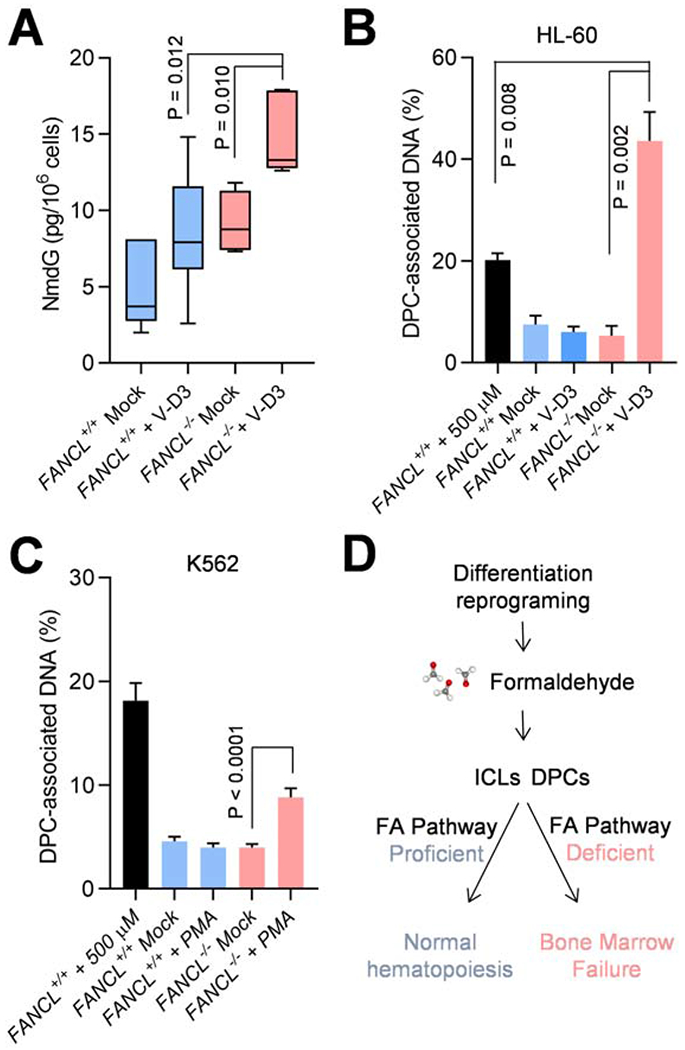
Impaired DNA crosslink repair leads to Fanconi anemia (FA), characterized by a unique manifestation of bone marrow failure and pancytopenia among diseases caused by DNA damage response defects. As a germline disorder, why the hematopoietic hierarchy is specifically affected is not fully understood. We find that reprogramming transcription during hematopoietic differentiation results in an overload of genotoxic stress, which causes aborted differentiation and depletion of FA mutant progenitor cells. DNA damage onset most likely arises from formaldehyde, an obligate by-product of oxidative protein demethylation during transcription regulation. Our results demonstrate that rapid and extensive transcription reprogramming associated with hematopoietic differentiation poses a major threat to genome stability and cell viability in the absence of the FA pathway. The connection between differentiation and DNA damage accumulation reveals a novel mechanism of genome scarring and is critical to exploring therapies to counteract the aplastic anemia for the treatment of FA patients.
Keywords: DNA damage; Fanconi anemia; bone marrow failure; differentiation; formaldehyde; hematopoiesis; transcription reprogramming.
Copyright © 2020 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of Interests The authors declare no conflict of interests.
Figures
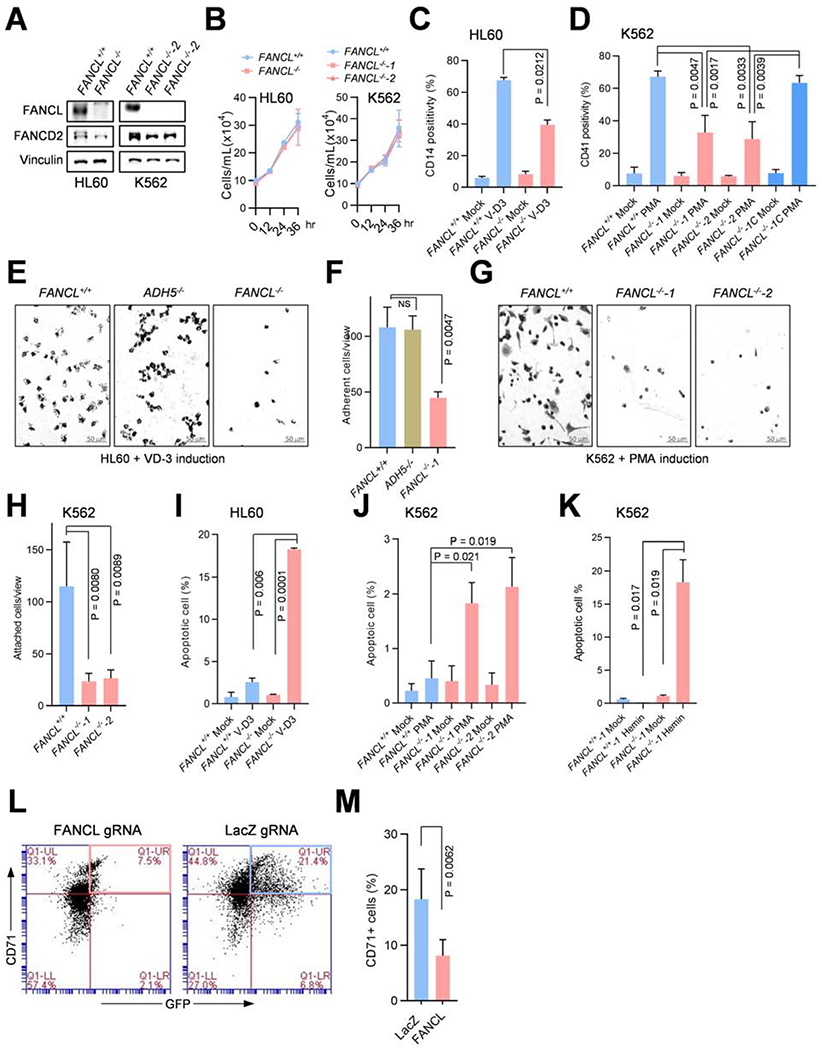
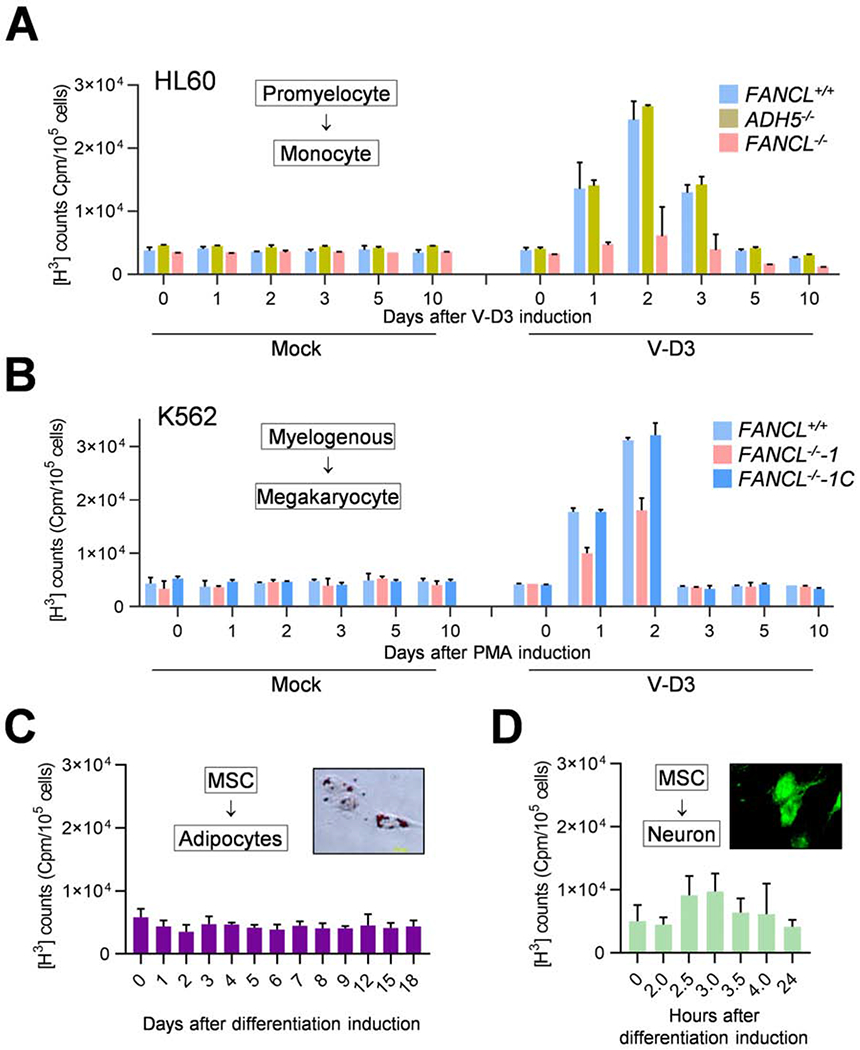
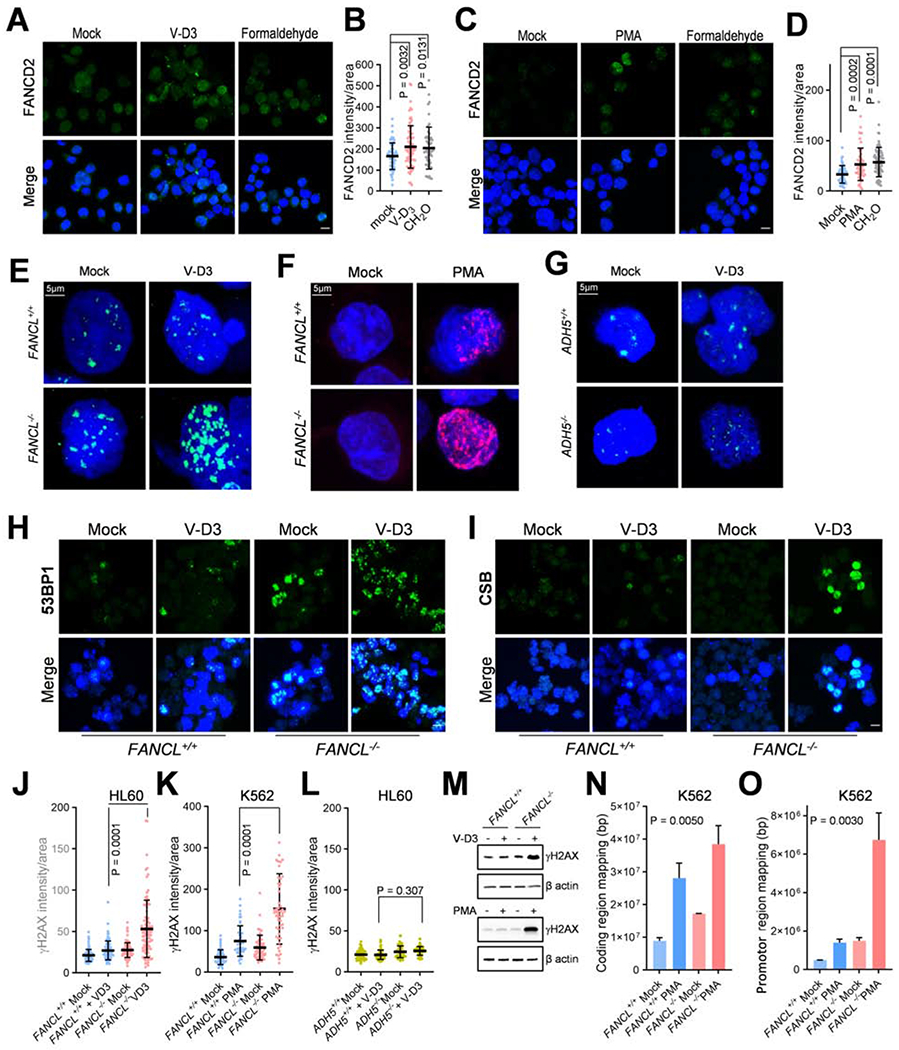
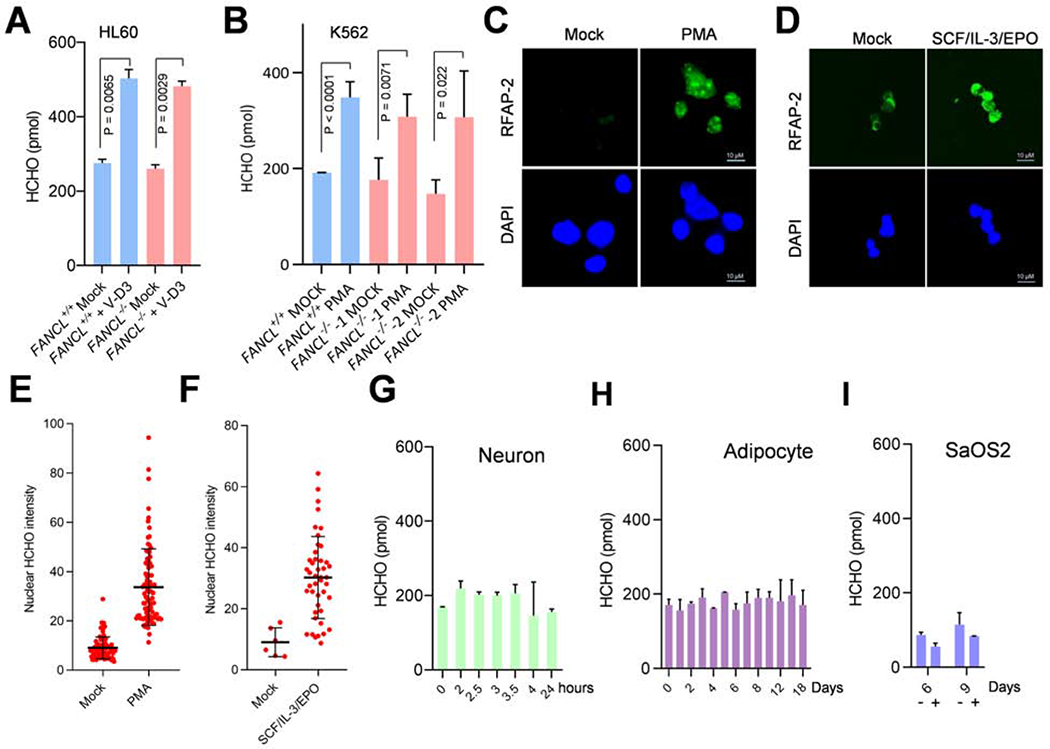
Comment in
-
Formaldehyde Causes Bone Marrow Failure Linked to Transcriptional Reprogramming or Metabolic Deficiency.Mol Cell. 2020 Dec 17;80(6):935-937. doi: 10.1016/j.molcel.2020.11.042. Mol Cell. 2020. PMID: 33338408
References
-
- Armstrong L, Stojkovic M, Dimmick I, Ahmad S, Stojkovic P, Hole N, and Lako M (2004). Phenotypic characterization of murine primitive hematopoietic progenitor cells isolated on basis of aldehyde dehydrogenase activity. Stem Cells 22, 1142–1151. - PubMed
-
- Ceccaldi R, Sarangi P, and D’Andrea AD (2016). The Fanconi anaemia pathway: new players and new functions. Nature Reviews Molecular Cell Biology 17, 337. - PubMed
-
- Costa M, Zhitkovich A, Gargas M, Paustenbach D, Finley B, Kuykendall J, Billings R, Carlson TJ, Wetterhahn K, Xu J, et al. (1996). Interlaboratory validation of a new assay for DNA-protein crosslinks. Mutat Res 369, 13–21. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
