

Submolecular probing of the complement C5a receptor-ligand binding reveals a cooperative two-site binding mechanism
- PMID: 33339958
- PMCID: PMC7749166
- DOI: 10.1038/s42003-020-01518-8
Submolecular probing of the complement C5a receptor-ligand binding reveals a cooperative two-site binding mechanism
Erratum in
-
Author Correction: Submolecular probing of the complement C5a receptor-ligand binding reveals a cooperative two-site binding mechanism.Commun Biol. 2021 May 12;4(1):592. doi: 10.1038/s42003-021-02174-2. Commun Biol. 2021. PMID: 33980997 Free PMC article. No abstract available.
Abstract
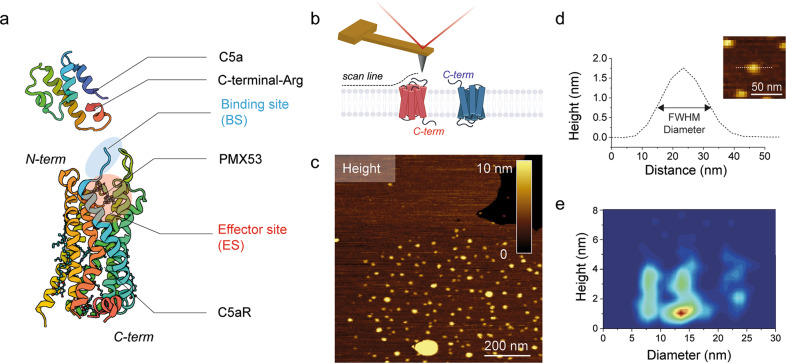
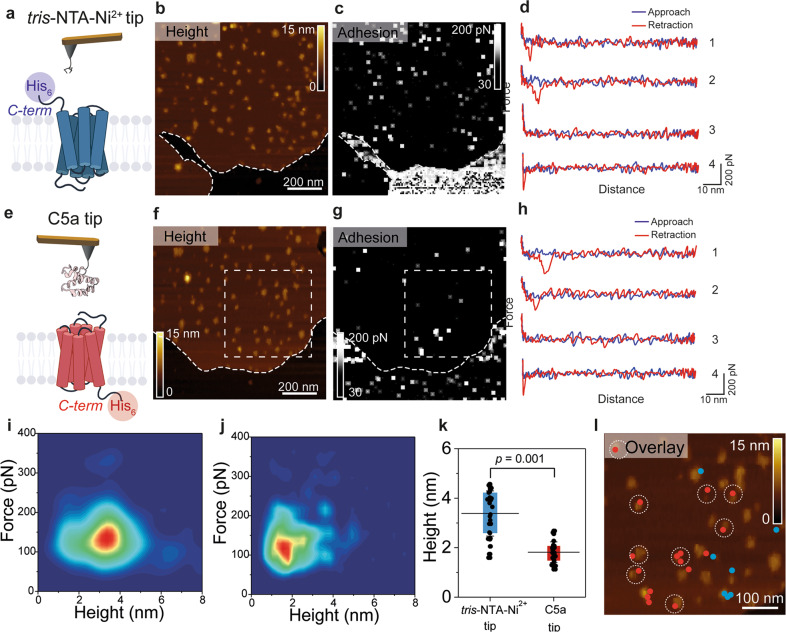
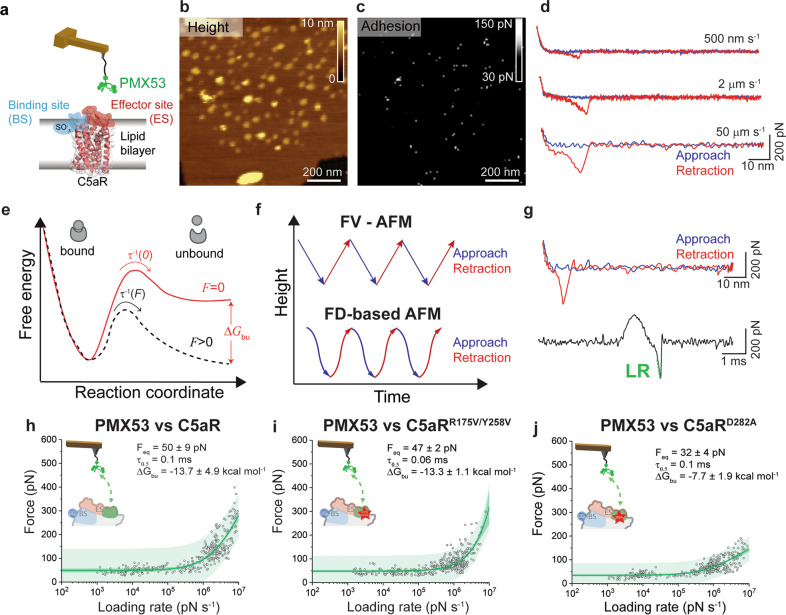
A current challenge to produce effective therapeutics is to accurately determine the location of the ligand-biding site and to characterize its properties. So far, the mechanisms underlying the functional activation of cell surface receptors by ligands with a complex binding mechanism remain poorly understood due to a lack of suitable nanoscopic methods to study them in their native environment. Here, we elucidated the ligand-binding mechanism of the human G protein-coupled C5a receptor (C5aR). We discovered for the first time a cooperativity between the two orthosteric binding sites. We found that the N-terminus C5aR serves as a kinetic trap, while the transmembrane domain acts as the functional site and both contributes to the overall high-affinity interaction. In particular, Asp282 plays a key role in ligand binding thermodynamics, as revealed by atomic force microscopy and steered molecular dynamics simulation. Our findings provide a new structural basis for the functional and mechanistic understanding of the GPCR family that binds large macromolecular ligands.
Conflict of interest statement
The authors declare no competing interests.
Figures



References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
