

Improving clinical trial outcomes in amyotrophic lateral sclerosis
- PMID: 33340024
- PMCID: PMC7747476
- DOI: 10.1038/s41582-020-00434-z
Improving clinical trial outcomes in amyotrophic lateral sclerosis
Abstract
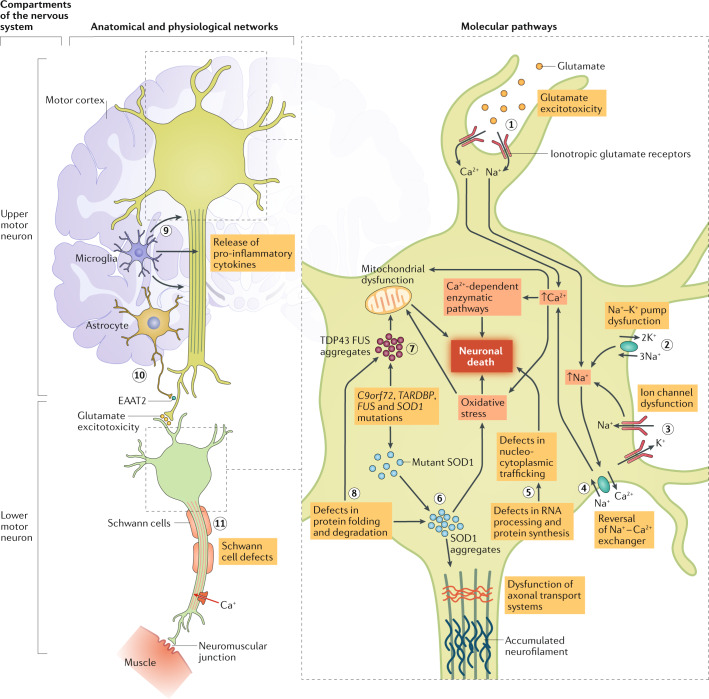
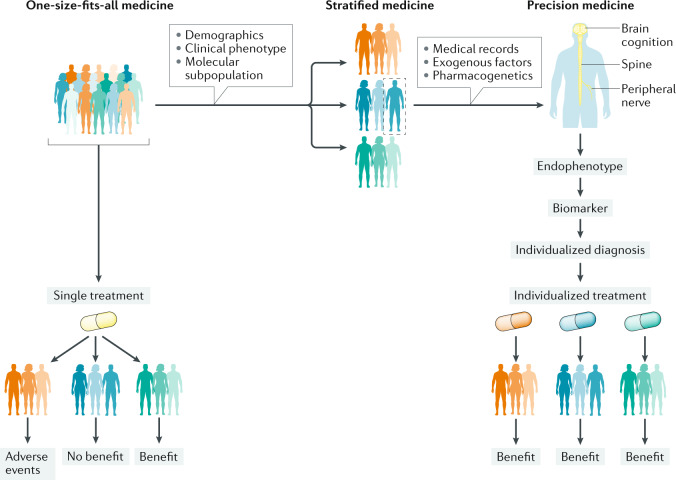
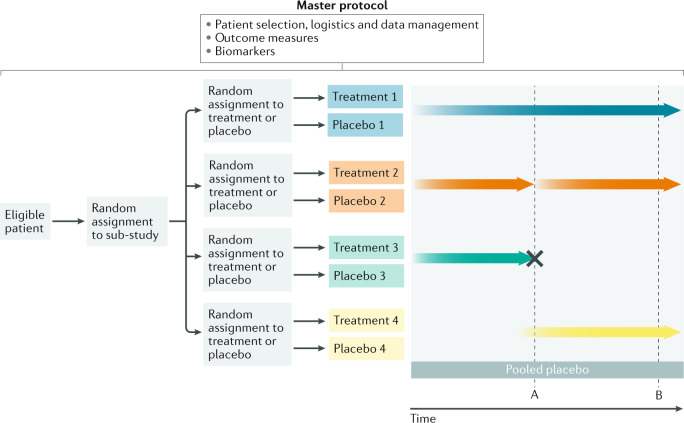
Individuals who are diagnosed with amyotrophic lateral sclerosis (ALS) today face the same historically intransigent problem that has existed since the initial description of the disease in the 1860s - a lack of effective therapies. In part, the development of new treatments has been hampered by an imperfect understanding of the biological processes that trigger ALS and promote disease progression. Advances in our understanding of these biological processes, including the causative genetic mutations, and of the influence of environmental factors have deepened our appreciation of disease pathophysiology. The consequent identification of pathogenic targets means that the introduction of effective therapies is becoming a realistic prospect. Progress in precision medicine, including genetically targeted therapies, will undoubtedly change the natural history of ALS. The evolution of clinical trial designs combined with improved methods for patient stratification will facilitate the translation of novel therapies into the clinic. In addition, the refinement of emerging biomarkers of therapeutic benefits is critical to the streamlining of care for individuals. In this Review, we synthesize these developments in ALS and discuss the further developments and refinements needed to accelerate the introduction of effective therapeutic approaches.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- Alzheimer’s Disease International. World Alzheimer report 2019: attitudes to dementia (ADI, 2019).
-
- World Health Organization. Global action plan on the public health response to dementia 2017–2025 (WHO, 2017).
Publication types
MeSH terms
Substances
Grants and funding
- MR/L501529/1/MRC_/Medical Research Council/United Kingdom
- TURNER/OCT18/989-797/MNDA_/Motor Neurone Disease Association/United Kingdom
- ALCHALABI-TALBOT/APR14/926-794/MNDA_/Motor Neurone Disease Association/United Kingdom
- MR/R024804/1/MRC_/Medical Research Council/United Kingdom
- WT_/Wellcome Trust/United Kingdom
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
