

The Neuromuscular Junction in Health and Disease: Molecular Mechanisms Governing Synaptic Formation and Homeostasis
- PMID: 33343299
- PMCID: PMC7744297
- DOI: 10.3389/fnmol.2020.610964
The Neuromuscular Junction in Health and Disease: Molecular Mechanisms Governing Synaptic Formation and Homeostasis
Abstract
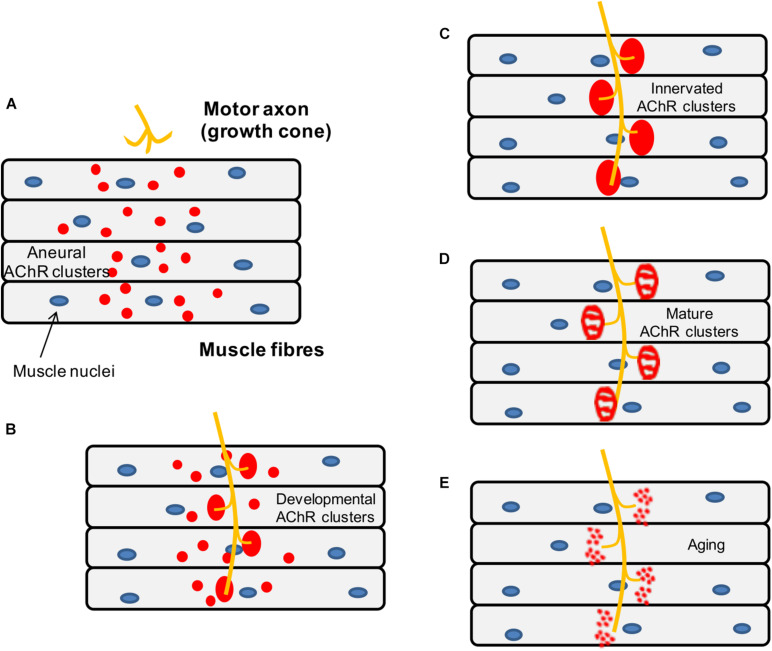
The neuromuscular junction (NMJ) is a highly specialized synapse between a motor neuron nerve terminal and its muscle fiber that are responsible for converting electrical impulses generated by the motor neuron into electrical activity in the muscle fibers. On arrival of the motor nerve action potential, calcium enters the presynaptic terminal, which leads to the release of the neurotransmitter acetylcholine (ACh). ACh crosses the synaptic gap and binds to ACh receptors (AChRs) tightly clustered on the surface of the muscle fiber; this leads to the endplate potential which initiates the muscle action potential that results in muscle contraction. This is a simplified version of the events in neuromuscular transmission that take place within milliseconds, and are dependent on a tiny but highly structured NMJ. Much of this review is devoted to describing in more detail the development, maturation, maintenance and regeneration of the NMJ, but first we describe briefly the most important molecules involved and the conditions that affect their numbers and function. Most important clinically worldwide, are myasthenia gravis (MG), the Lambert-Eaton myasthenic syndrome (LEMS) and congenital myasthenic syndromes (CMS), each of which causes specific molecular defects. In addition, we mention the neurotoxins from bacteria, snakes and many other species that interfere with neuromuscular transmission and cause potentially fatal diseases, but have also provided useful probes for investigating neuromuscular transmission. There are also changes in NMJ structure and function in motor neuron disease, spinal muscle atrophy and sarcopenia that are likely to be secondary but might provide treatment targets. The NMJ is one of the best studied and most disease-prone synapses in the nervous system and it is amenable to in vivo and ex vivo investigation and to systemic therapies that can help restore normal function.
Keywords: Agrin; DOK7; MuSK; congenital myasthenic syndromes; myasthenia gravis; neuromuscular junction; sarcopenia; spinal muscular atrophy.
Copyright © 2020 Rodríguez Cruz, Cossins, Beeson and Vincent.
Conflict of interest statement
University of Oxford and AV hold patents and receive royalties for antibody assays from Euroimmun AG and Athena Diagnostics. The remaining authors declare that the literature review was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
References
-
- Akaaboune M., Culican S. M., Turney S. G., Jeff W., Akaaboune M., Culican S. M., et al. (1999). Rapid and reversible effects of activity on acetylcholine receptor density at the neuromuscular junction in vivo. Science 286 503–507. - PubMed
-
- Akaaboune M., Grady R. M., Turney S., Sanes J. R., Lichtman J. W., Louis S. (2002). Neurotechnique dynamics studied in vivo by reversible photo-unbinding of fluorescent ligands. Neuron 34 865–876. - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous