

Early life imprints the hierarchy of T cell clone sizes
- PMID: 33345776
- PMCID: PMC7870140
- DOI: 10.7554/eLife.61639
Early life imprints the hierarchy of T cell clone sizes
Abstract
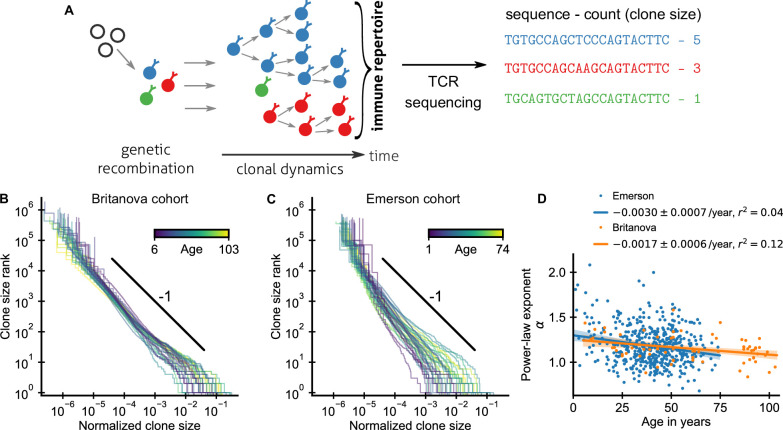
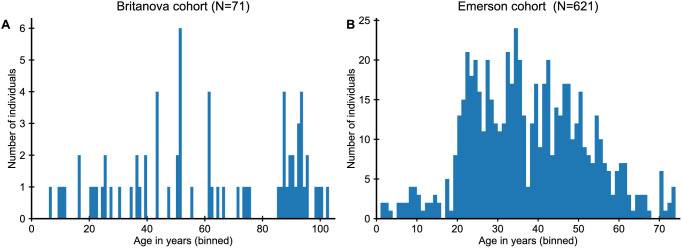
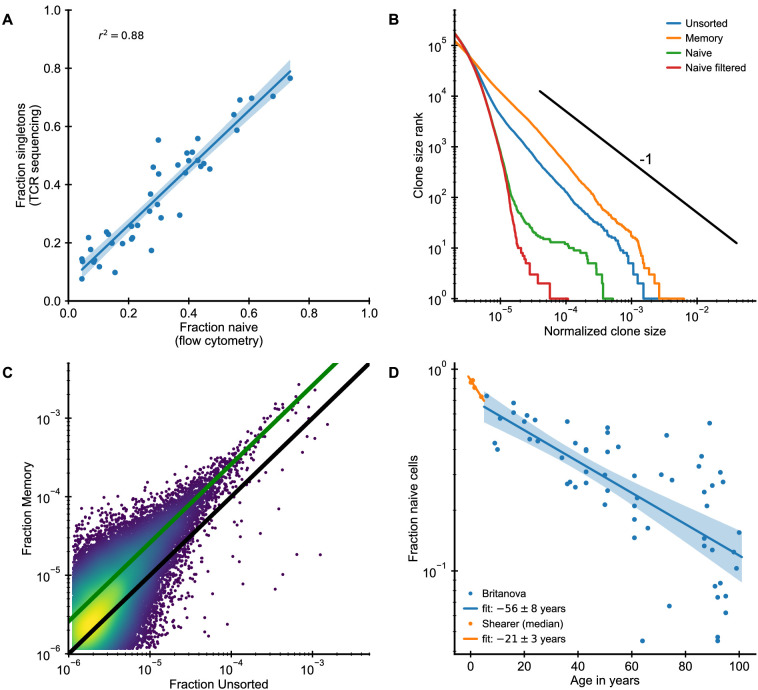
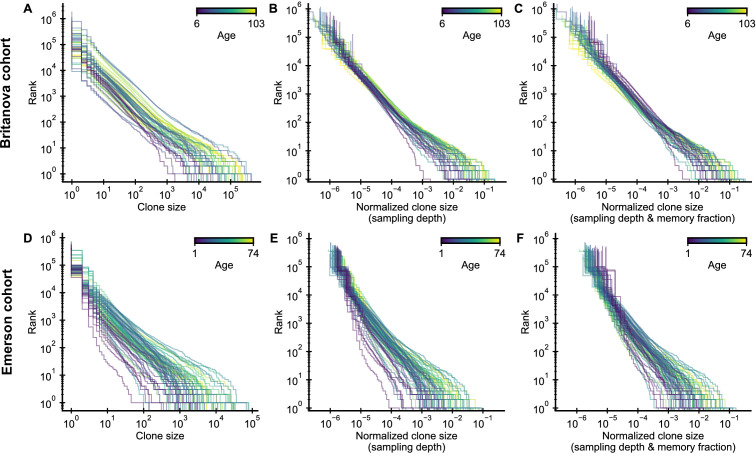
The adaptive immune system responds to pathogens by selecting clones of cells with specific receptors. While clonal selection in response to particular antigens has been studied in detail, it is unknown how a lifetime of exposures to many antigens collectively shape the immune repertoire. Here, using mathematical modeling and statistical analyses of T cell receptor sequencing data, we develop a quantitative theory of human T cell dynamics compatible with the statistical laws of repertoire organization. We find that clonal expansions during a perinatal time window leave a long-lasting imprint on the human T cell repertoire, which is only slowly reshaped by fluctuating clonal selection during adult life. Our work provides a mechanism for how early clonal dynamics imprint the hierarchy of T cell clone sizes with implications for pathogen defense and autoimmunity.
Keywords: T cell immunity; fluctuating fitness; high-dimensional ecology; human; immune repertoire; immunology; imprinting; inflammation; physics of living systems; power-law scaling; repertoire sequencing.
Plain language summary
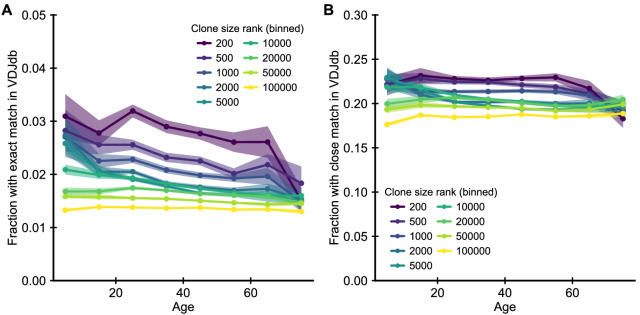
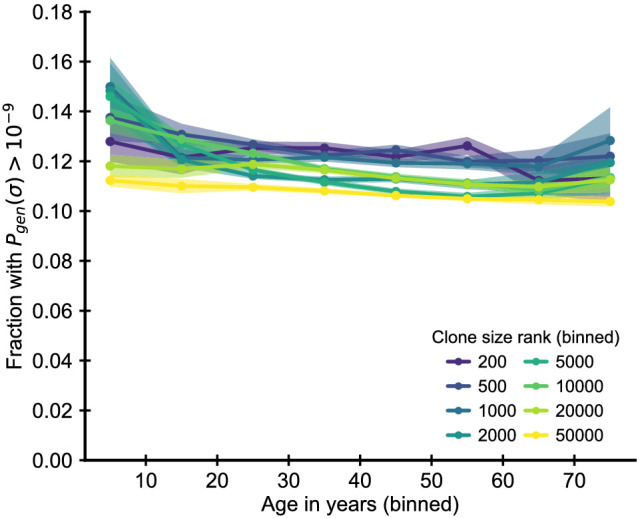
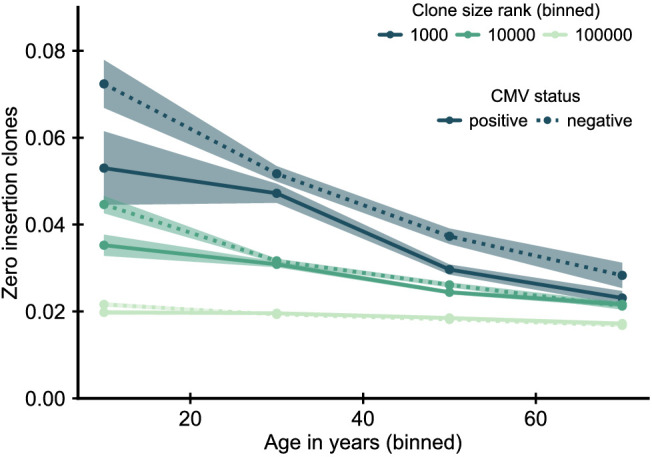
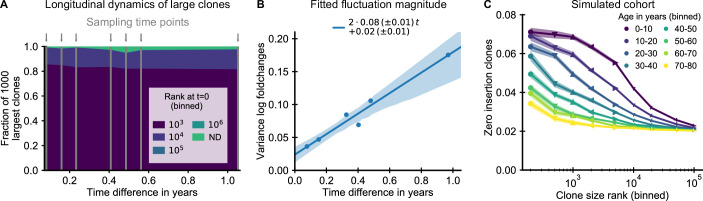
The human immune system develops a memory of pathogens that it encounters over its lifetime, allowing it to respond quickly to future infections. It does this partly through T cells, white blood cells that can recognize different pathogens. During an infection, the T cells that recognize the specific pathogen attacking the body will divide until a large number of clones of these T cells is available to help in the fight. After the infection clears, the immune system ‘keeps’ some of these cells so it can recognize the pathogen in the future, and respond quicker to an infection. Over the course of their lives, people will be infected by many different pathogens, leading to a wide variety of T cells that each respond to one of these pathogens. However, it is not well understood how various infections throughout the human lifespan shape the overall population of different T cells. Gaimann et al. used mathematical modelling to study how the composition of the immune system changes in people of different ages. Different populations of T cells – each specialized against a specific antigen – had been previously identified through genetic sequencing. Gaimann et al. analyzed their dynamics to show that many of the largest populations originate around birth, during the formation of the immune system. These findings suggest a potential mechanism for how exposure to pathogens in infancy can influence the immune system much later in life. The results may also explain variations in how people respond to infections and in their risk of developing autoimmune conditions. This understanding could help develop new treatments or interventions to guide the immune system as it develops.
© 2020, Gaimann et al.
Conflict of interest statement
MG, MN, JD, AM No competing interests declared
Figures
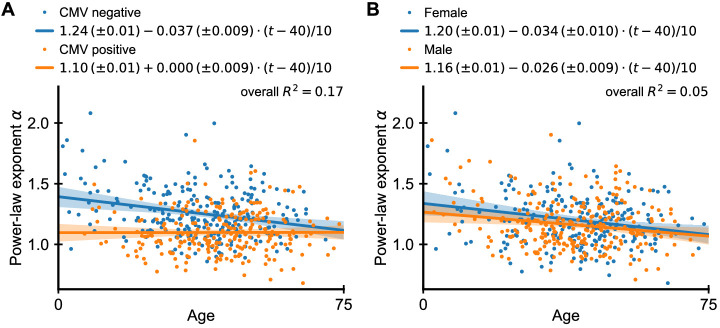
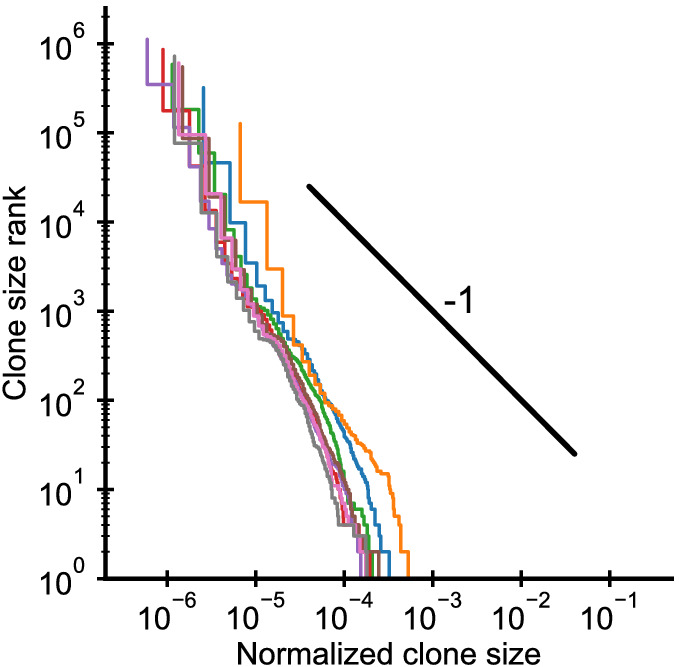
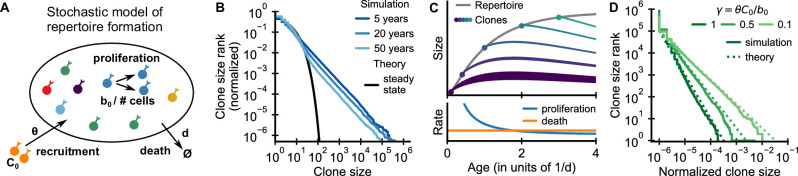
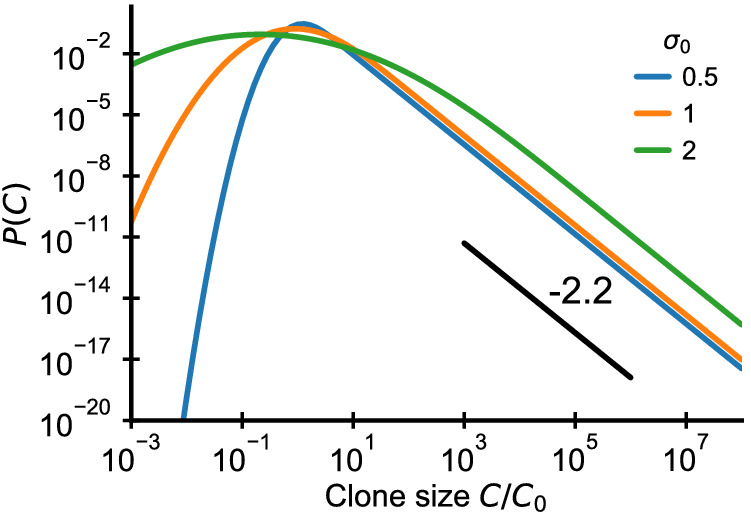
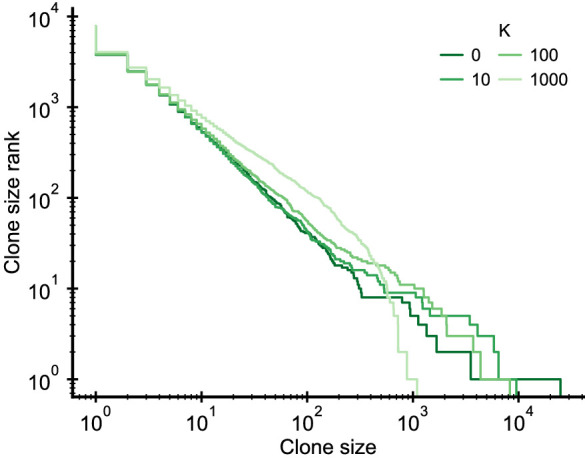
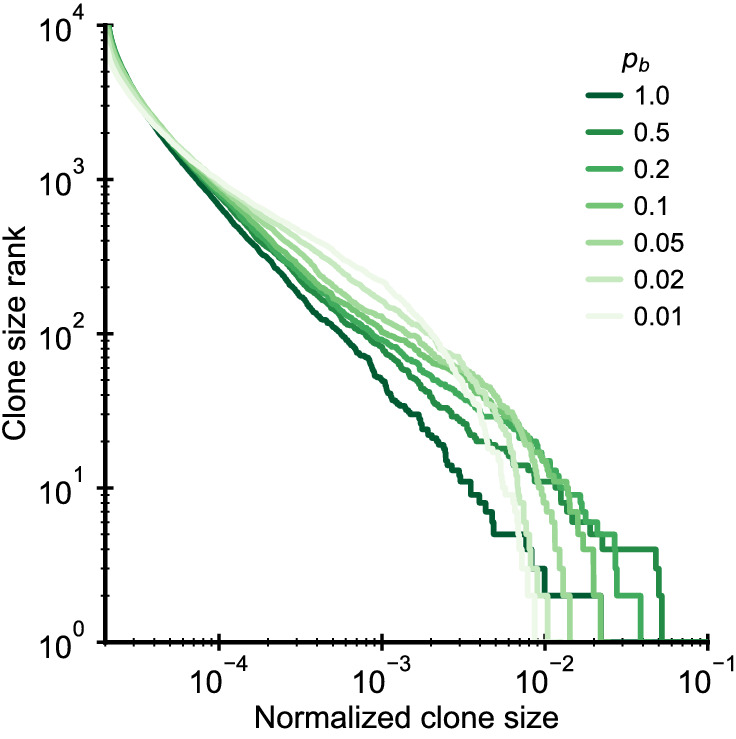
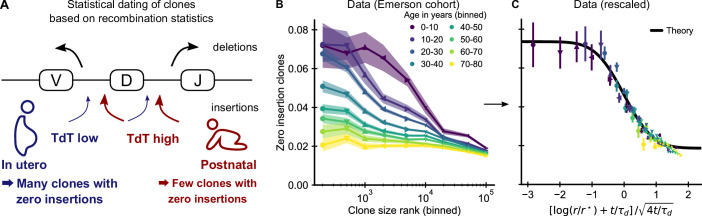
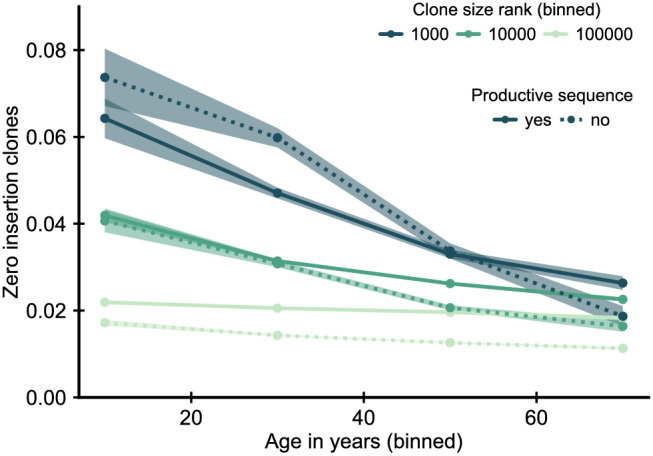
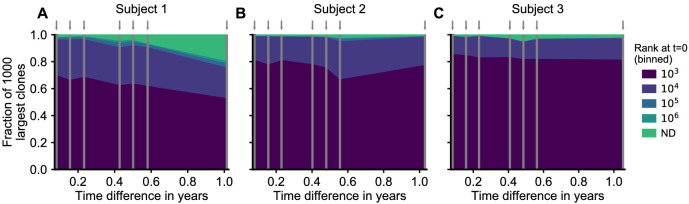
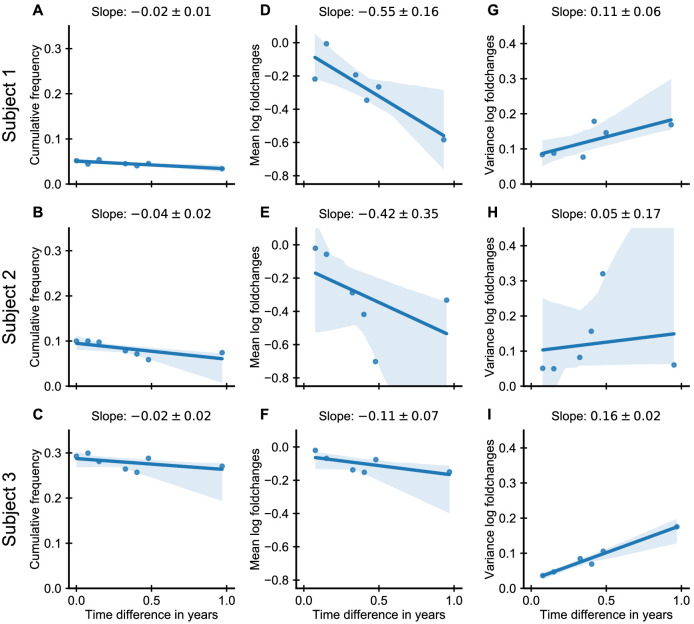
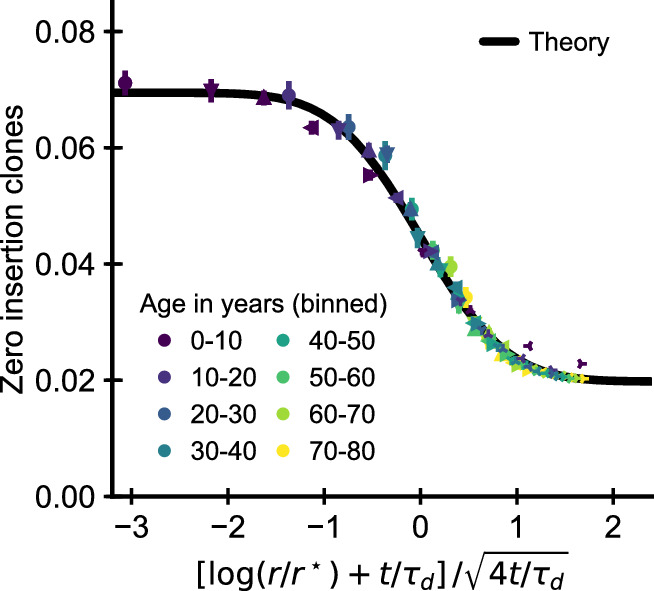
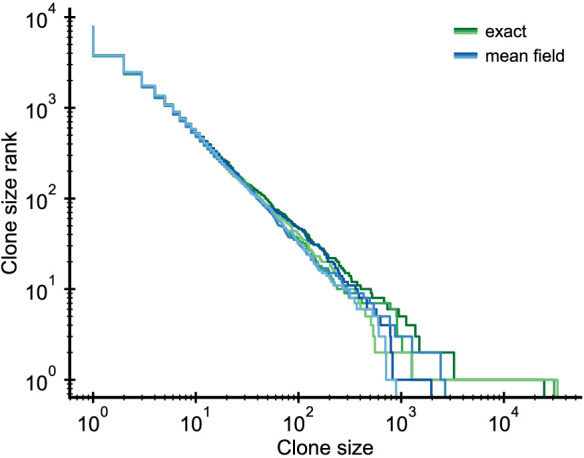
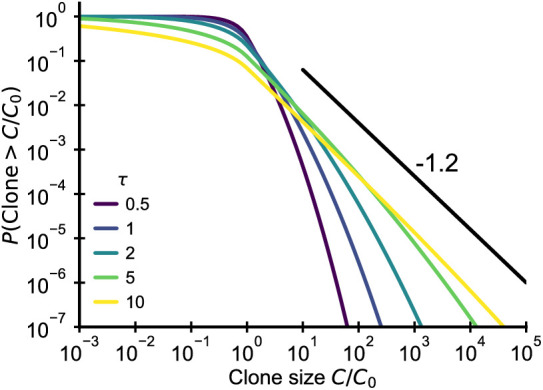
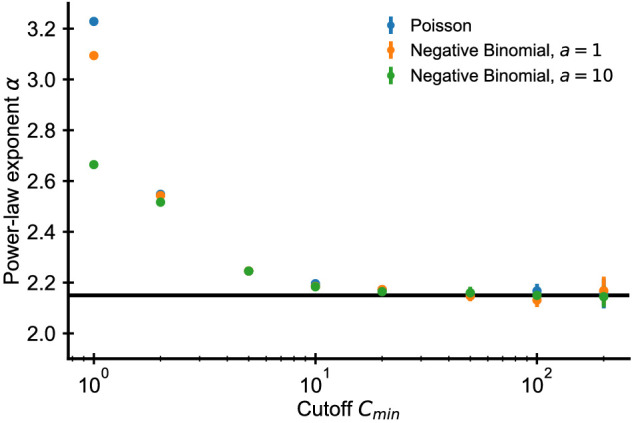
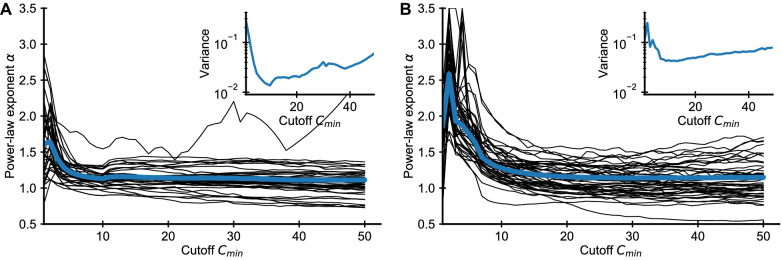
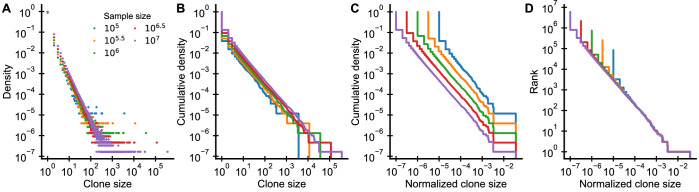
References
-
- Akondy RS, Fitch M, Edupuganti S, Yang S, Kissick HT, Li KW, Youngblood BA, Abdelsamed HA, McGuire DJ, Cohen KW, Alexe G, Nagar S, McCausland MM, Gupta S, Tata P, Haining WN, McElrath MJ, Zhang D, Hu B, Greenleaf WJ, Goronzy JJ, Mulligan MJ, Hellerstein M, Ahmed R. Origin and differentiation of human memory CD8 T cells after vaccination. Nature. 2017;552:362–367. doi: 10.1038/nature24633. - DOI - PMC - PubMed
-
- Altan-Bonnet G, Mora T, Walczak AM. Quantitative immunology for physicists. Physics Reports. 2020;849:1–83. doi: 10.1016/j.physrep.2020.01.001. - DOI
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
