

Multi-omics data integration considerations and study design for biological systems and disease
- PMID: 33347526
- PMCID: PMC8058243
- DOI: 10.1039/d0mo00041h
Multi-omics data integration considerations and study design for biological systems and disease
Abstract
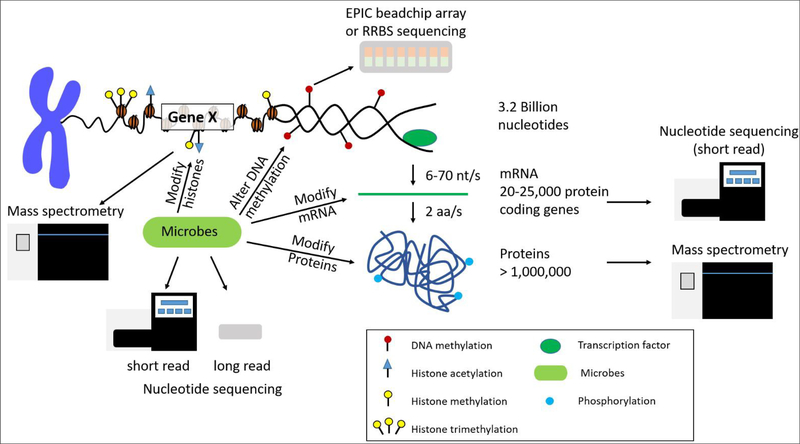
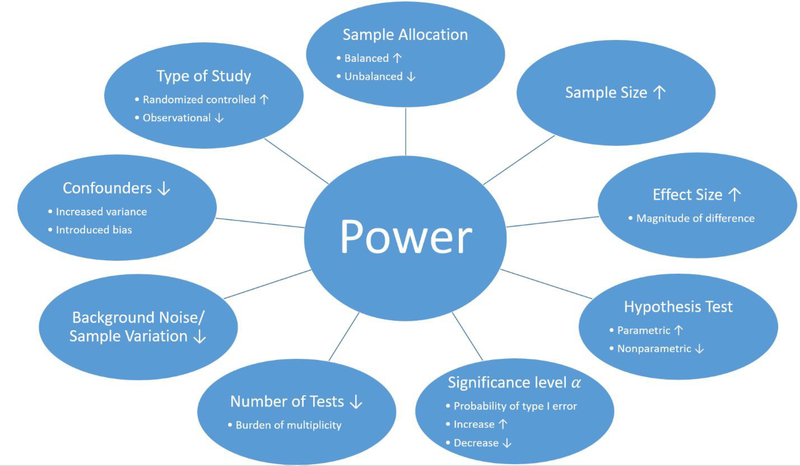
With the advancement of next-generation sequencing and mass spectrometry, there is a growing need for the ability to merge biological features in order to study a system as a whole. Features such as the transcriptome, methylome, proteome, histone post-translational modifications and the microbiome all influence the host response to various diseases and cancers. Each of these platforms have technological limitations due to sample preparation steps, amount of material needed for sequencing, and sequencing depth requirements. These features provide a snapshot of one level of regulation in a system. The obvious next step is to integrate this information and learn how genes, proteins, and/or epigenetic factors influence the phenotype of a disease in context of the system. In recent years, there has been a push for the development of data integration methods. Each method specifically integrates a subset of omics data using approaches such as conceptual integration, statistical integration, model-based integration, networks, and pathway data integration. In this review, we discuss considerations of the study design for each data feature, the limitations in gene and protein abundance and their rate of expression, the current data integration methods, and microbiome influences on gene and protein expression. The considerations discussed in this review should be regarded when developing new algorithms for integrating multi-omics data.
Conflict of interest statement
Conflicts of interest
No potential conflict of interest was reported by the authors.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources