

Stepwise evolution of Salmonella Typhimurium ST313 causing bloodstream infection in Africa
- PMID: 33349664
- PMCID: PMC8018540
- DOI: 10.1038/s41564-020-00836-1
Stepwise evolution of Salmonella Typhimurium ST313 causing bloodstream infection in Africa
Abstract
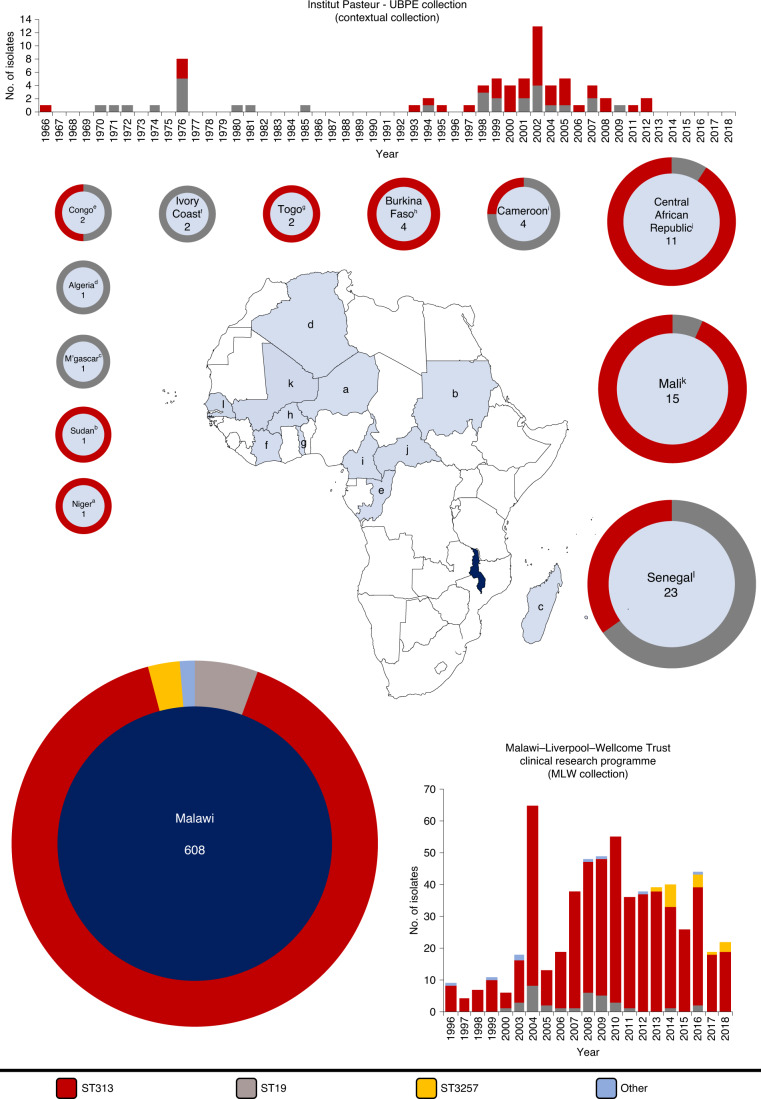
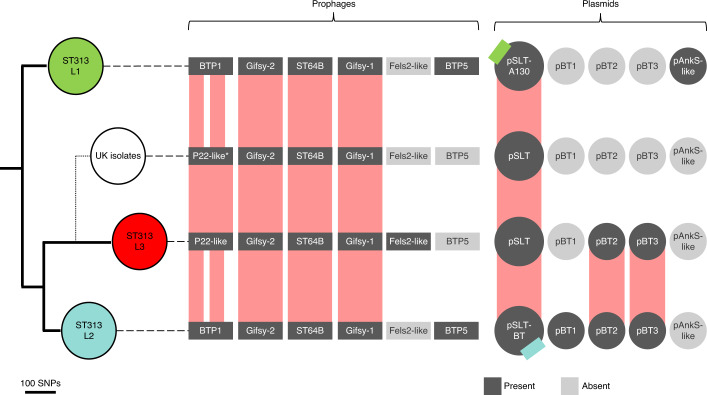
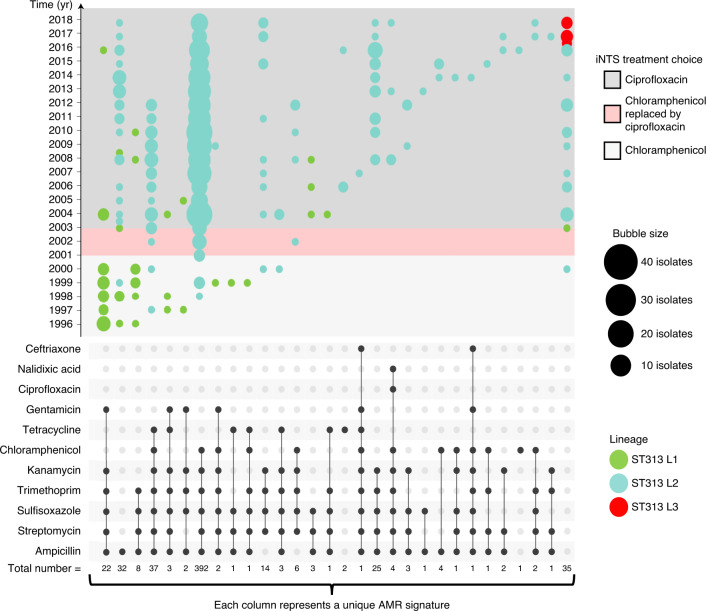
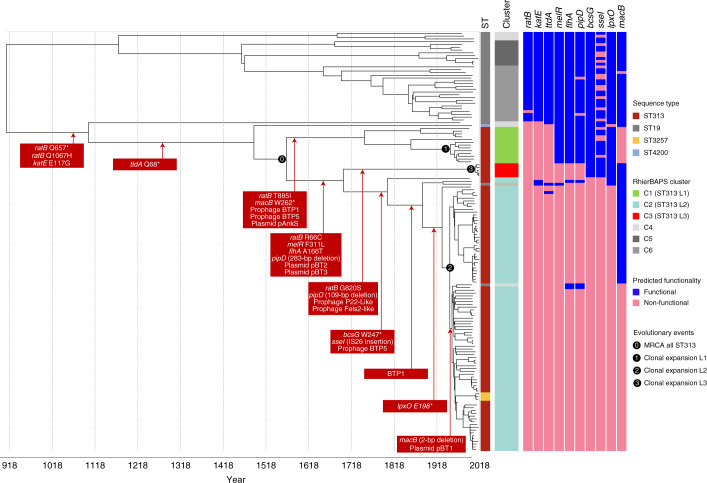
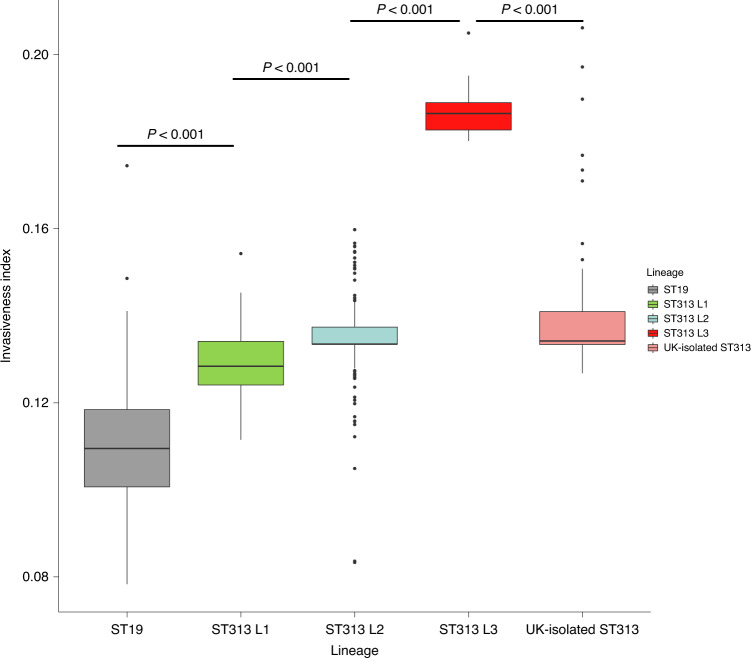
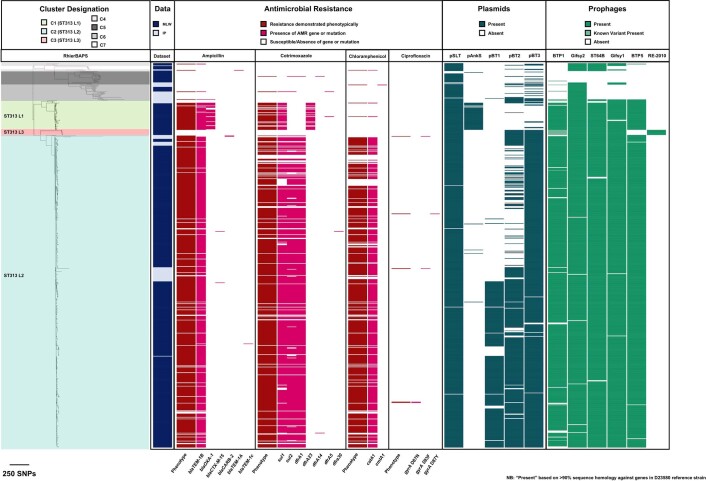
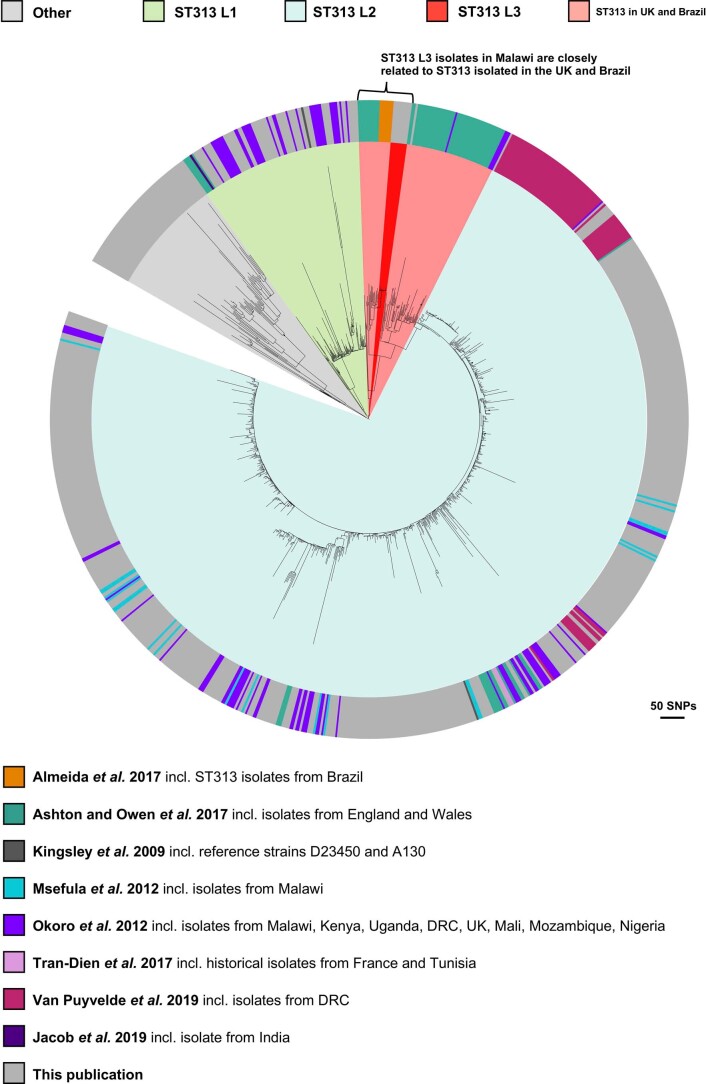
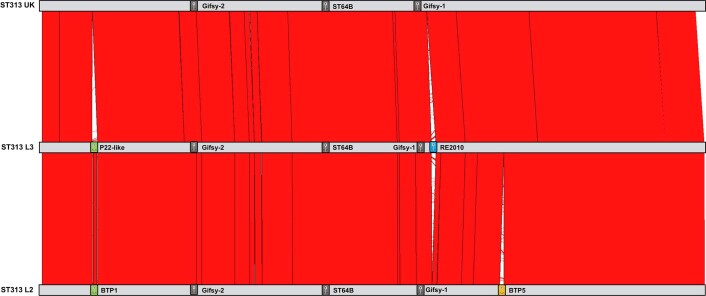
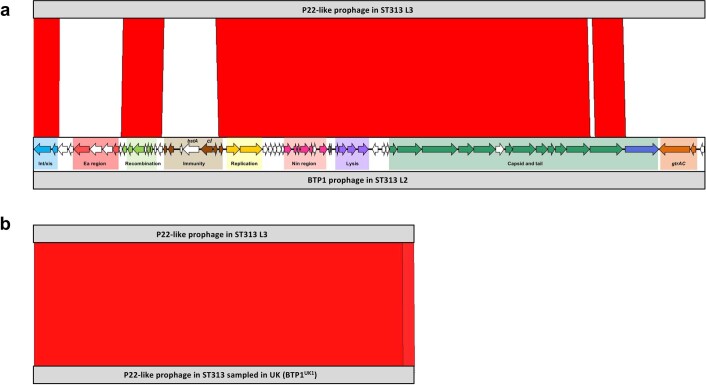
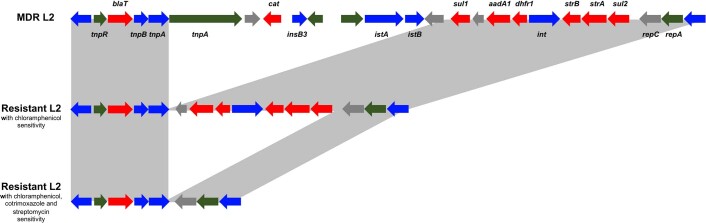
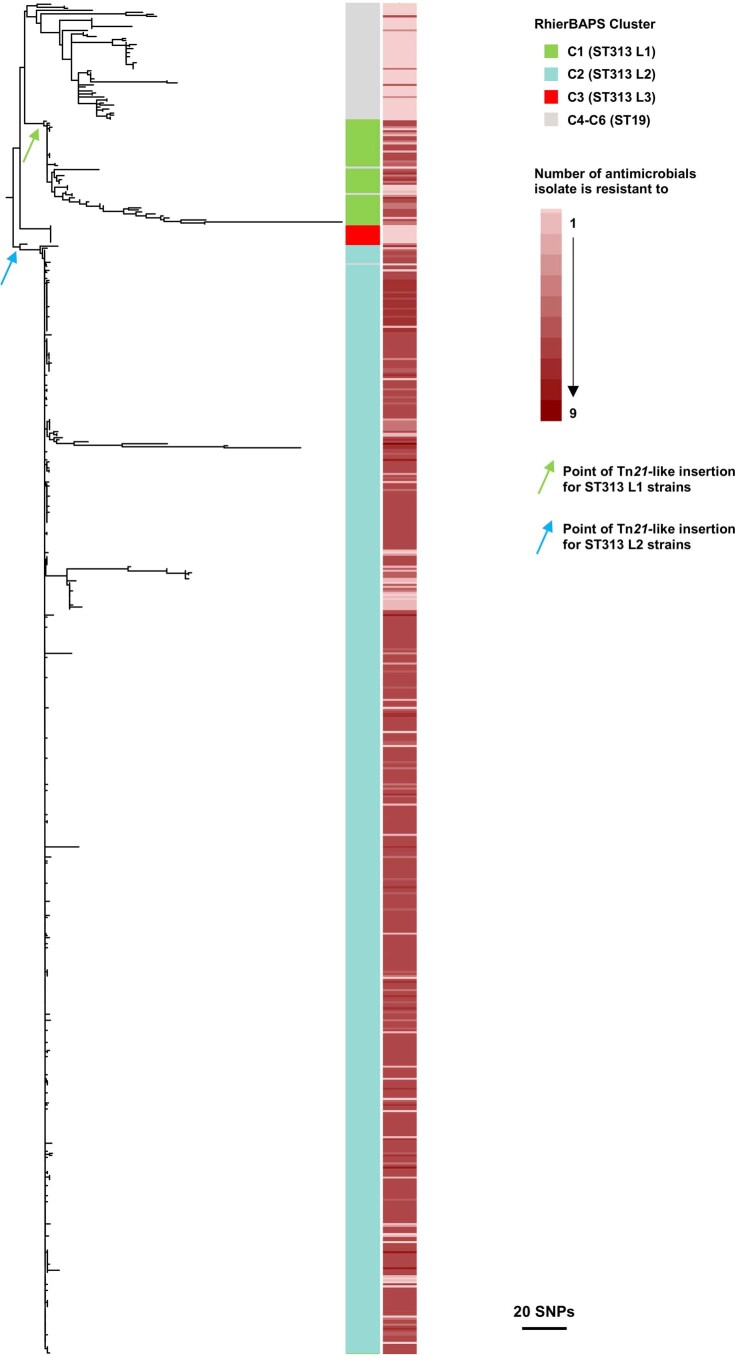
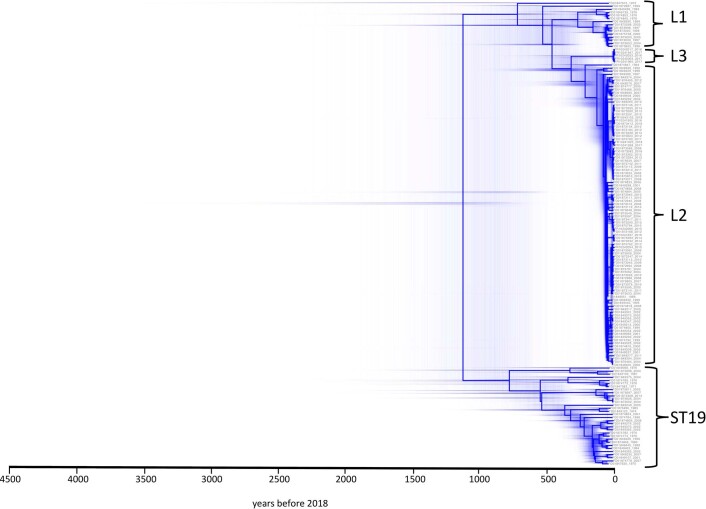
Bloodstream infections caused by nontyphoidal Salmonella are a major public health concern in Africa, causing ~49,600 deaths every year. The most common Salmonella enterica pathovariant associated with invasive nontyphoidal Salmonella disease is Salmonella Typhimurium sequence type (ST)313. It has been proposed that antimicrobial resistance and genome degradation has contributed to the success of ST313 lineages in Africa, but the evolutionary trajectory of such changes was unclear. Here, to define the evolutionary dynamics of ST313, we sub-sampled from two comprehensive collections of Salmonella isolates from African patients with bloodstream infections, spanning 1966 to 2018. The resulting 680 genome sequences led to the discovery of a pan-susceptible ST313 lineage (ST313 L3), which emerged in Malawi in 2016 and is closely related to ST313 variants that cause gastrointestinal disease in the United Kingdom and Brazil. Genomic analysis revealed degradation events in important virulence genes in ST313 L3, which had not occurred in other ST313 lineages. Despite arising only recently in the clinic, ST313 L3 is a phylogenetic intermediate between ST313 L1 and L2, with a characteristic accessory genome. Our in-depth genotypic and phenotypic characterization identifies the crucial loss-of-function genetic events that occurred during the stepwise evolution of invasive S. Typhimurium across Africa.
Conflict of interest statement
R.C. was employed by the University of Liverpool at the time of the study and is now an employee of the GSK group of companies. The remaining authors declare no competing interests.
Figures

Comment in
-
Emergence of invasive Salmonella in Africa.Nat Microbiol. 2021 Mar;6(3):273-274. doi: 10.1038/s41564-021-00864-5. Nat Microbiol. 2021. PMID: 33510475 No abstract available.
References
Publication types
MeSH terms
Grants and funding
- MR/R020787/1/MRC_/Medical Research Council/United Kingdom
- BB/CCG1720/1/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- BB/CSP17270/1/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- BBS/OS/GC/000009D/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- 106914/WT_/Wellcome Trust/United Kingdom
LinkOut - more resources
Full Text Sources
Medical
