

A multicenter study benchmarking single-cell RNA sequencing technologies using reference samples
- PMID: 33349700
- PMCID: PMC11245320
- DOI: 10.1038/s41587-020-00748-9
A multicenter study benchmarking single-cell RNA sequencing technologies using reference samples
Abstract
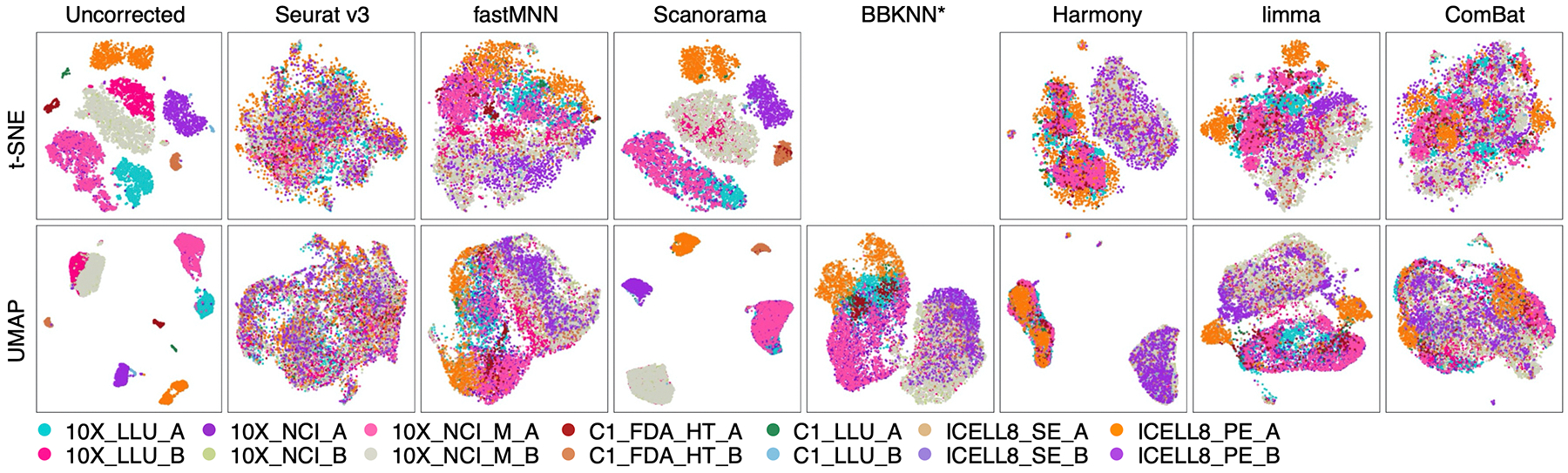
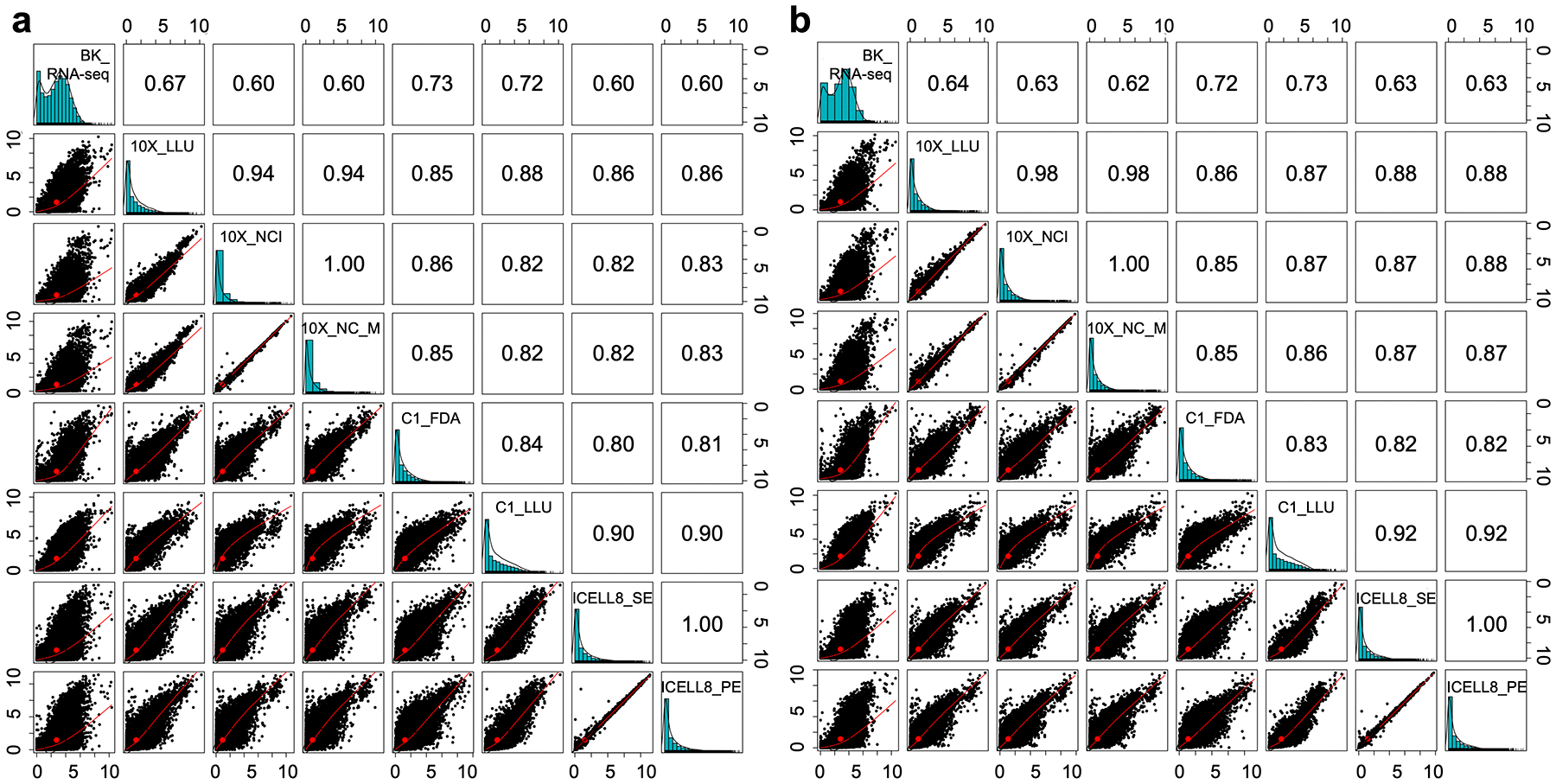
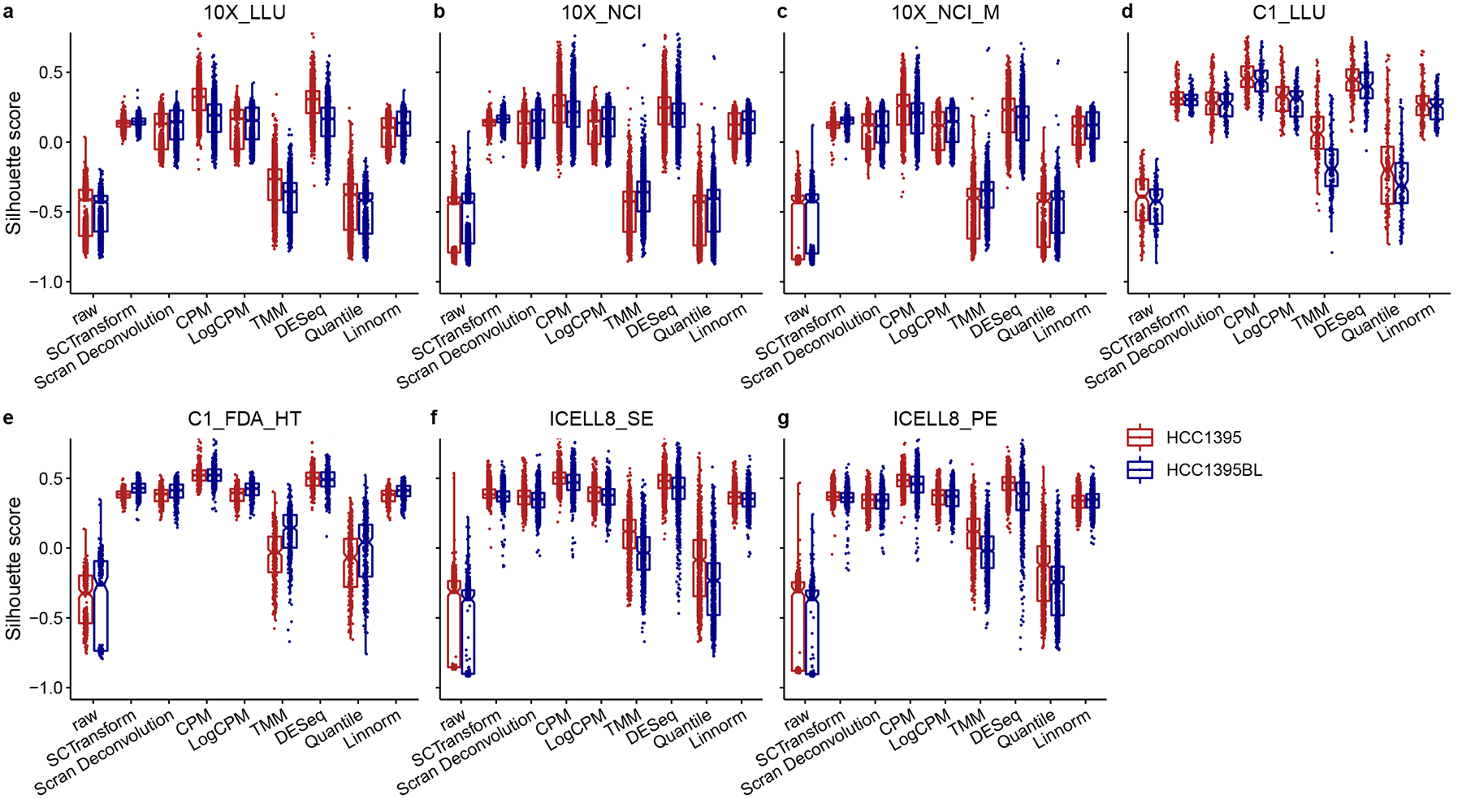
Comparing diverse single-cell RNA sequencing (scRNA-seq) datasets generated by different technologies and in different laboratories remains a major challenge. Here we address the need for guidance in choosing algorithms leading to accurate biological interpretations of varied data types acquired with different platforms. Using two well-characterized cellular reference samples (breast cancer cells and B cells), captured either separately or in mixtures, we compared different scRNA-seq platforms and several preprocessing, normalization and batch-effect correction methods at multiple centers. Although preprocessing and normalization contributed to variability in gene detection and cell classification, batch-effect correction was by far the most important factor in correctly classifying the cells. Moreover, scRNA-seq dataset characteristics (for example, sample and cellular heterogeneity and platform used) were critical in determining the optimal bioinformatic method. However, reproducibility across centers and platforms was high when appropriate bioinformatic methods were applied. Our findings offer practical guidance for optimizing platform and software selection when designing an scRNA-seq study.
© 2020. The Author(s), under exclusive licence to Springer Nature America, Inc.
Conflict of interest statement
Competing interests statement
Andrew Farmer and Alain Mir are employees of Takara Bio USA, Inc., and Ben Ernest and Urvashi Mehra were employees of Digicon Corporation. All other authors claim no conflicts of interest. The views presented in this article do not necessarily reflect current or future opinion or policy of the US Food and Drug Administration. Any mention of commercial products is for clarification and not intended as an endorsement.
Figures
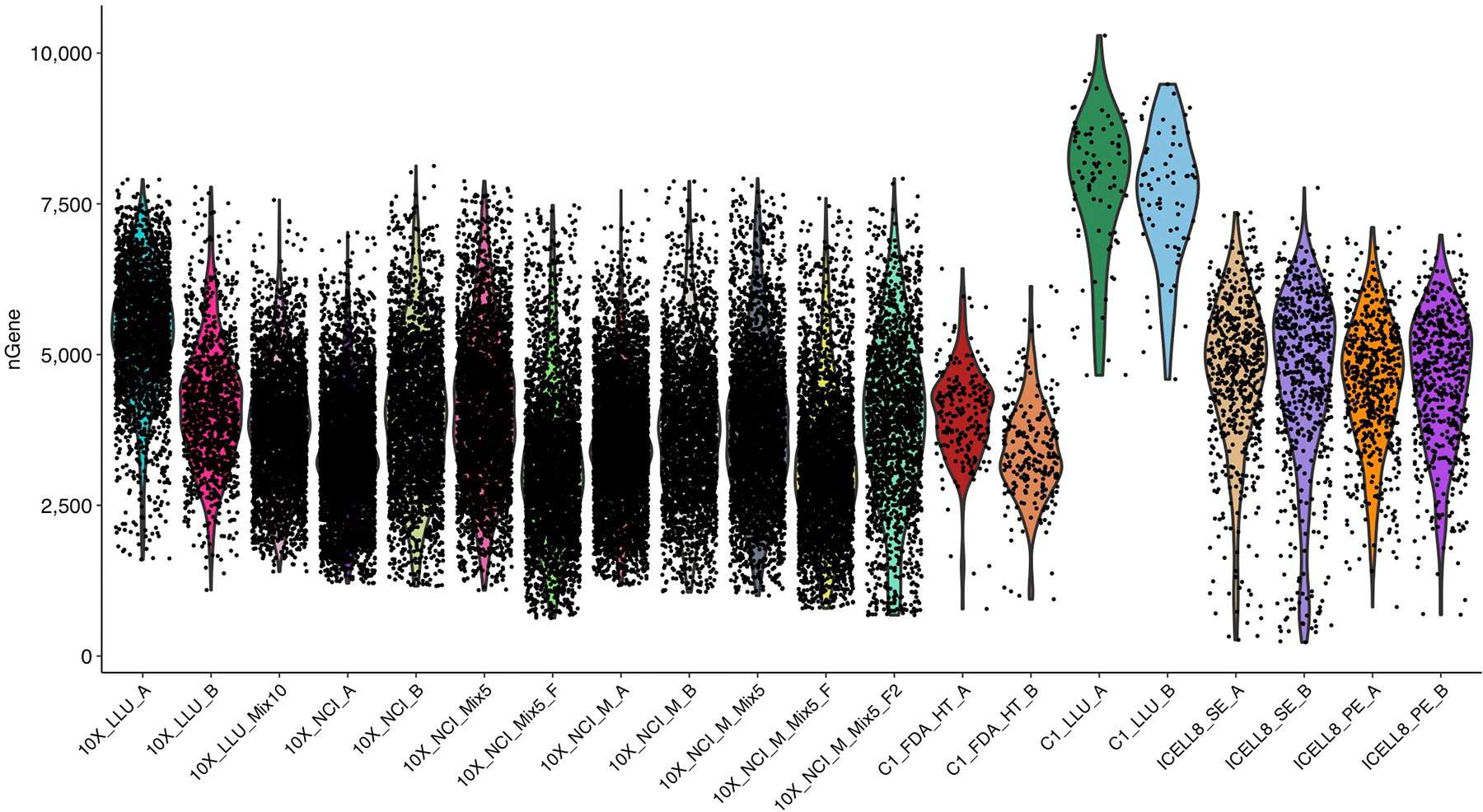
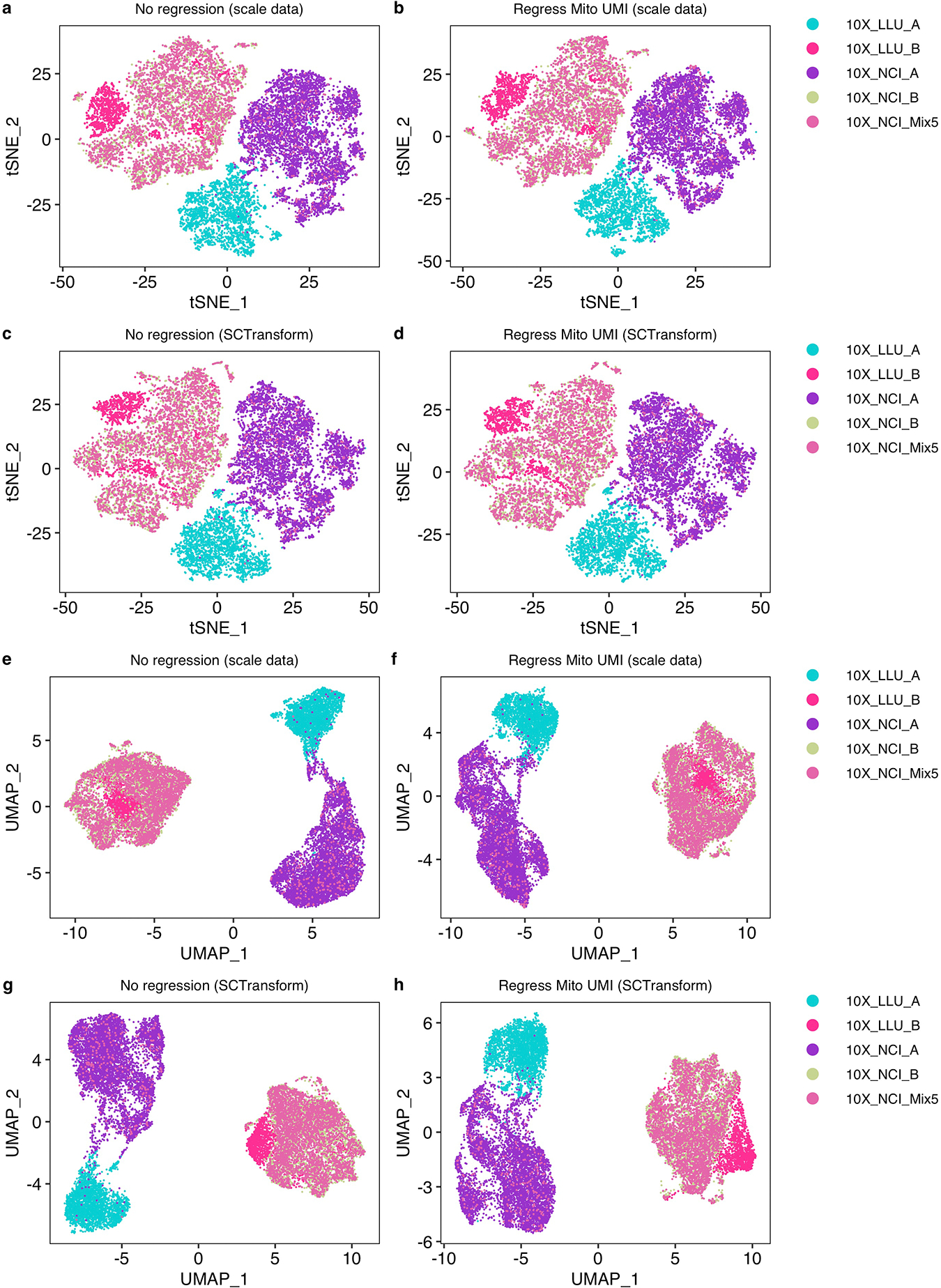

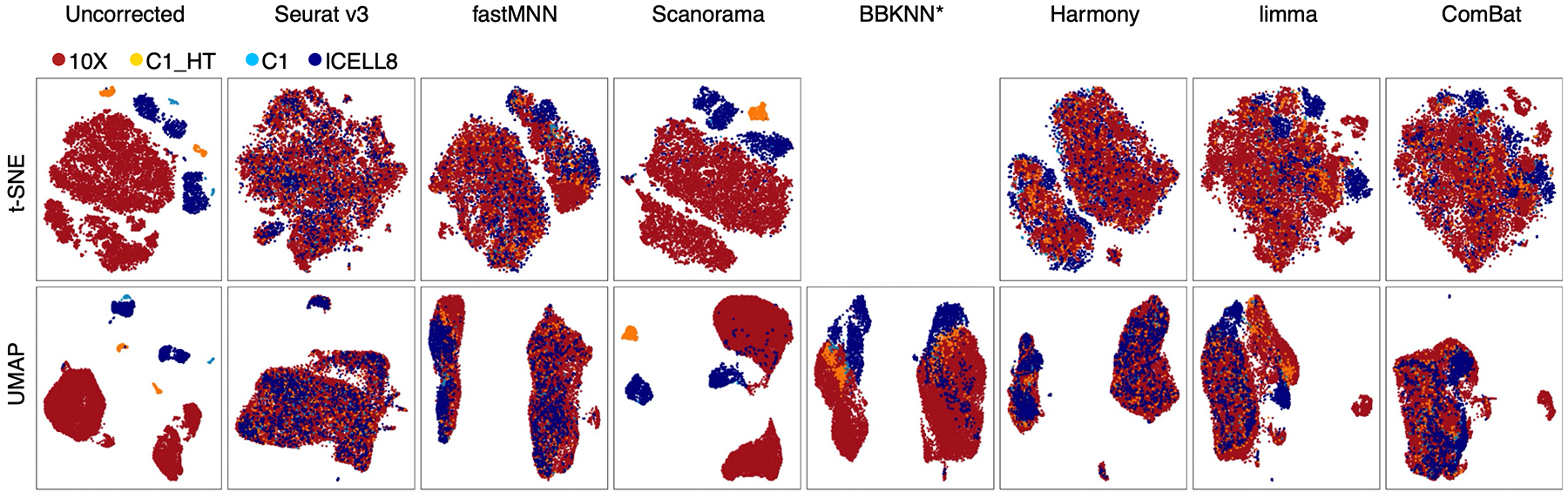
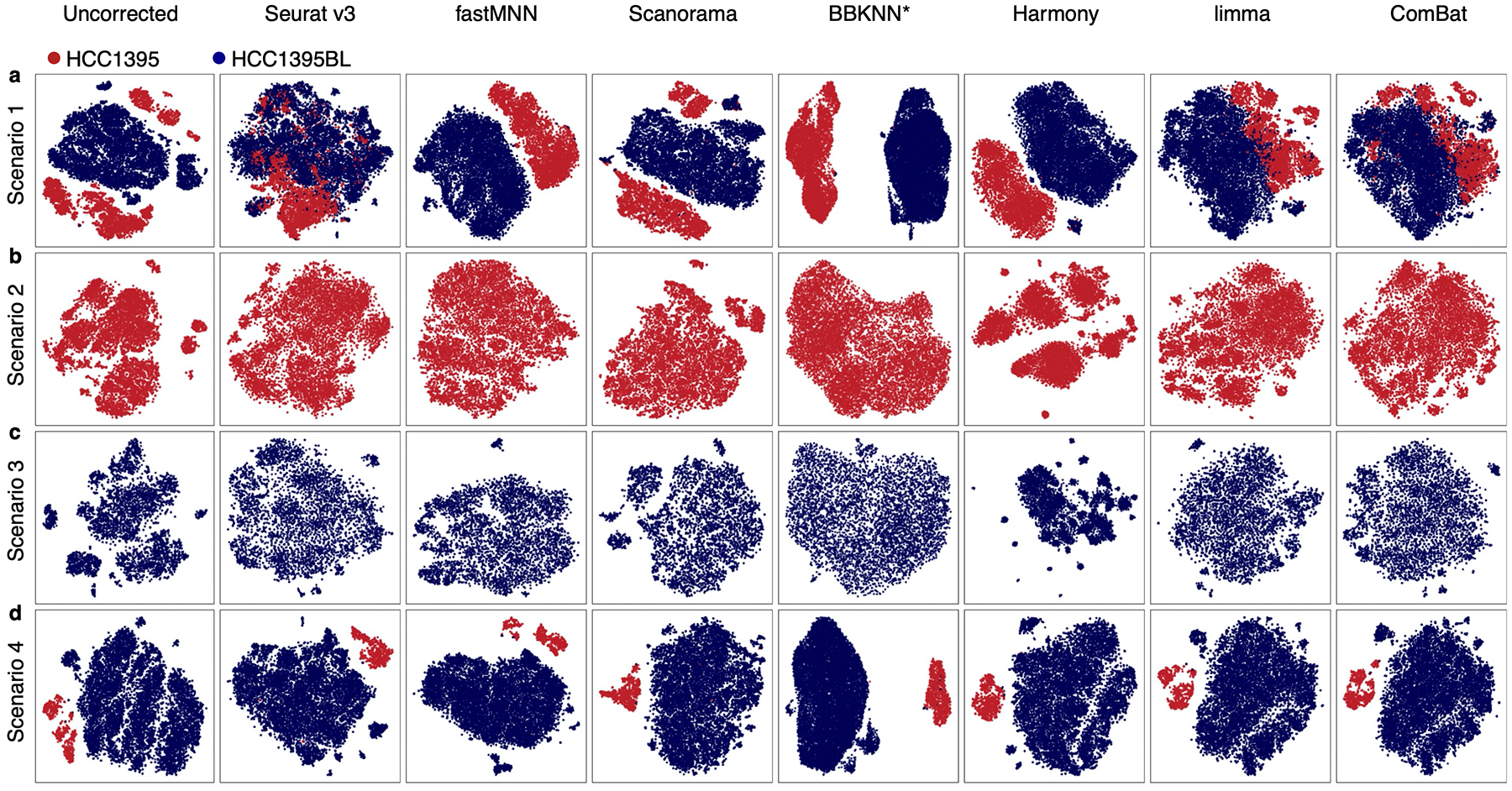








Similar articles
-
A multi-center cross-platform single-cell RNA sequencing reference dataset.Sci Data. 2021 Feb 2;8(1):39. doi: 10.1038/s41597-021-00809-x. Sci Data. 2021. PMID: 33531477 Free PMC article.
-
Benchmarking Algorithms for Gene Set Scoring of Single-cell ATAC-seq Data.Genomics Proteomics Bioinformatics. 2024 Jul 3;22(2):qzae014. doi: 10.1093/gpbjnl/qzae014. Genomics Proteomics Bioinformatics. 2024. PMID: 39049508 Free PMC article.
-
Data normalization for addressing the challenges in the analysis of single-cell transcriptomic datasets.BMC Genomics. 2024 May 6;25(1):444. doi: 10.1186/s12864-024-10364-5. BMC Genomics. 2024. PMID: 38711017 Free PMC article. Review.
-
Benchmarking UMI-based single-cell RNA-seq preprocessing workflows.Genome Biol. 2021 Dec 14;22(1):339. doi: 10.1186/s13059-021-02552-3. Genome Biol. 2021. PMID: 34906205 Free PMC article.
-
A Single-Cell Sequencing Guide for Immunologists.Front Immunol. 2018 Oct 23;9:2425. doi: 10.3389/fimmu.2018.02425. eCollection 2018. Front Immunol. 2018. PMID: 30405621 Free PMC article. Review.
Cited by
-
Personalized genome assembly for accurate cancer somatic mutation discovery using tumor-normal paired reference samples.Genome Biol. 2022 Nov 9;23(1):237. doi: 10.1186/s13059-022-02803-x. Genome Biol. 2022. PMID: 36352452 Free PMC article.
-
Systematic comparison of sequencing-based spatial transcriptomic methods.Nat Methods. 2024 Sep;21(9):1743-1754. doi: 10.1038/s41592-024-02325-3. Epub 2024 Jul 4. Nat Methods. 2024. PMID: 38965443 Free PMC article.
-
Reference Materials for Improving Reliability of Multiomics Profiling.Phenomics. 2024 Mar 6;4(5):487-521. doi: 10.1007/s43657-023-00153-7. eCollection 2024 Oct. Phenomics. 2024. PMID: 39723231 Free PMC article. Review.
-
Translational opportunities of single-cell biology in atherosclerosis.Eur Heart J. 2023 Apr 7;44(14):1216-1230. doi: 10.1093/eurheartj/ehac686. Eur Heart J. 2023. PMID: 36478058 Free PMC article. Review.
-
Sequence modification on demand: search and replace tools for precise gene editing in plants.Transgenic Res. 2021 Aug;30(4):353-379. doi: 10.1007/s11248-021-00253-y. Epub 2021 Jun 4. Transgenic Res. 2021. PMID: 34086167 Review.
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
