

Microglia facilitate repair of demyelinated lesions via post-squalene sterol synthesis
- PMID: 33349711
- PMCID: PMC7116742
- DOI: 10.1038/s41593-020-00757-6
Microglia facilitate repair of demyelinated lesions via post-squalene sterol synthesis
Abstract
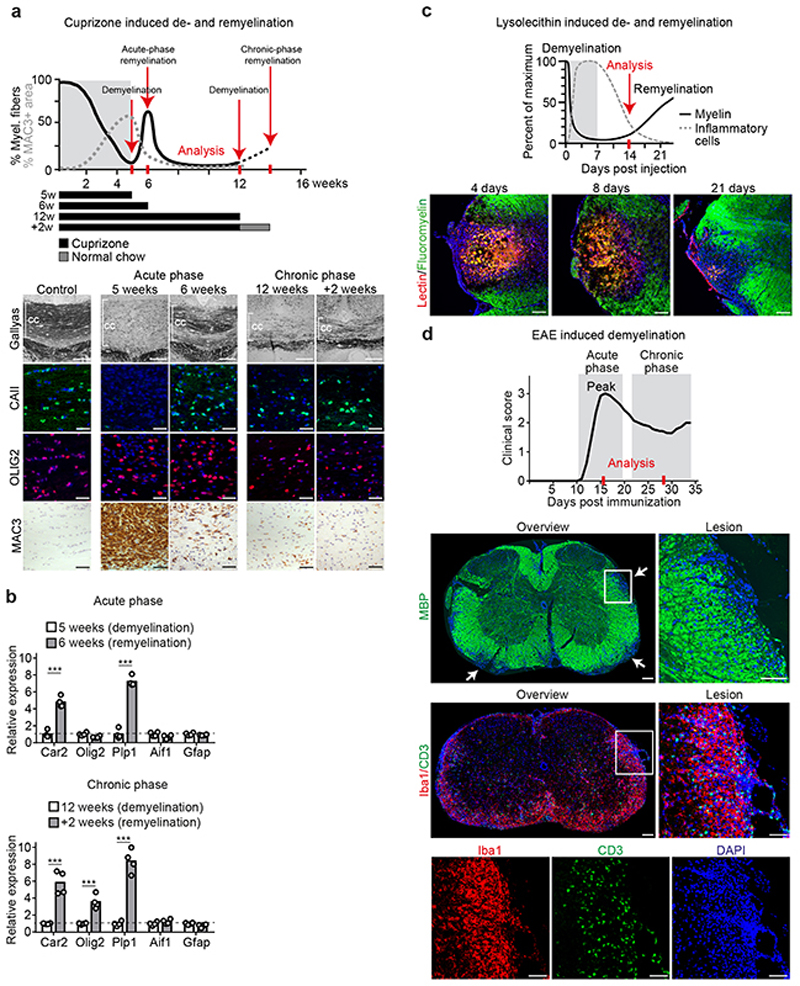
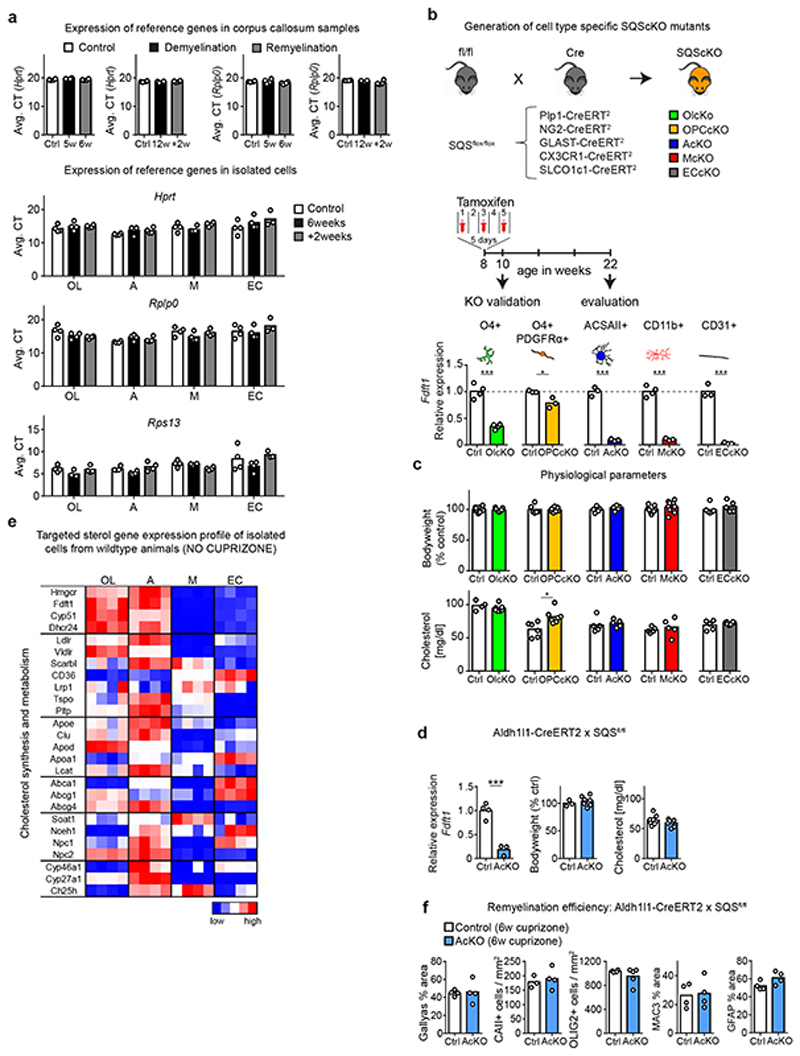
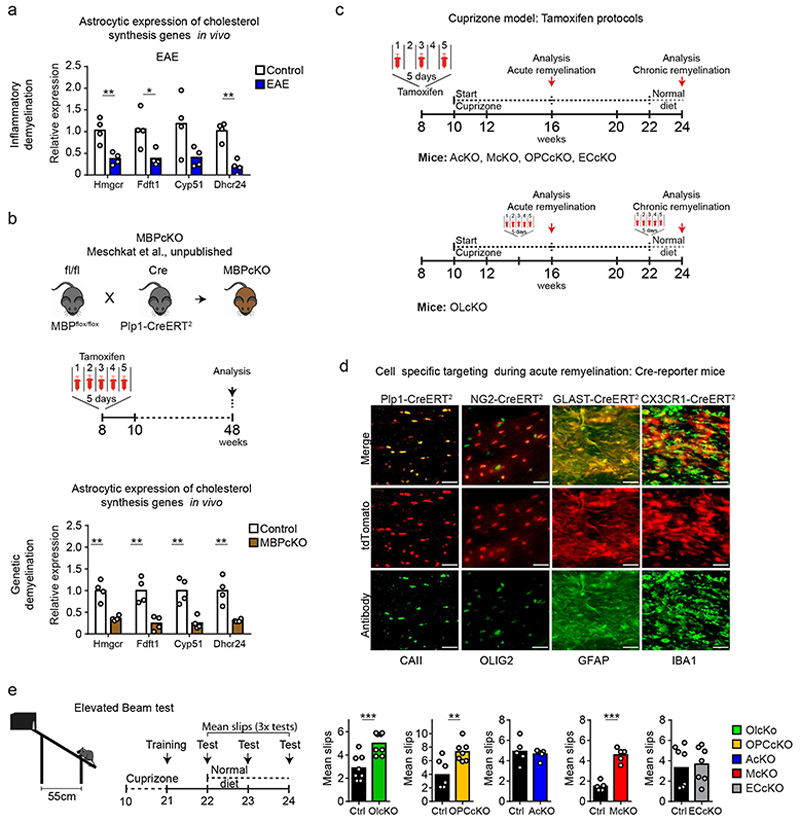
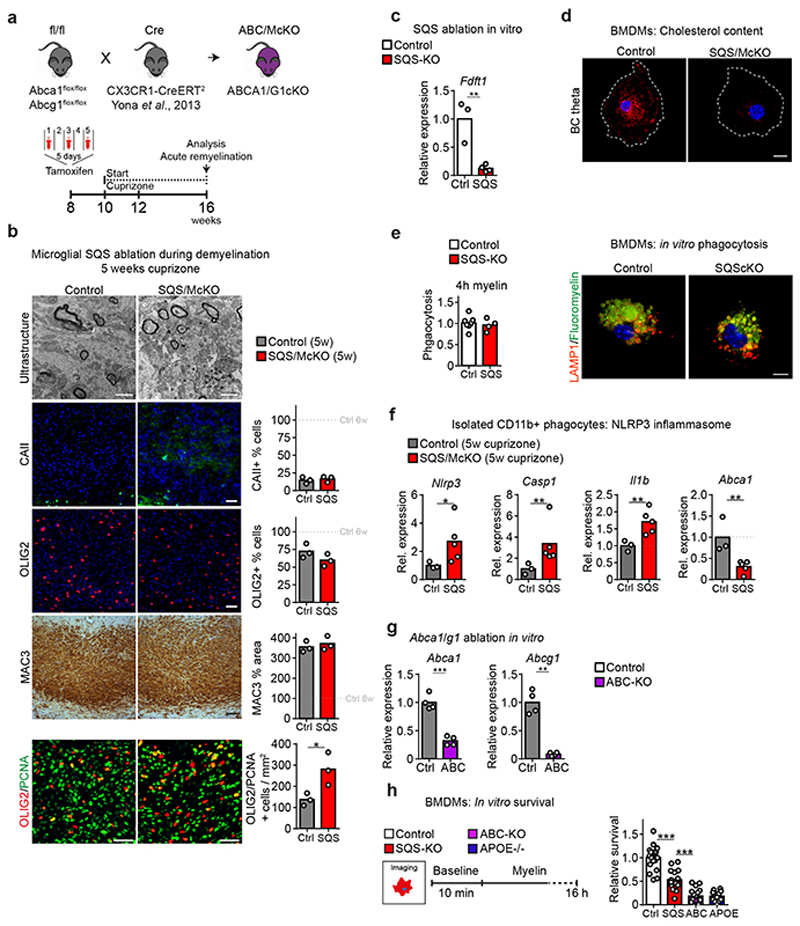
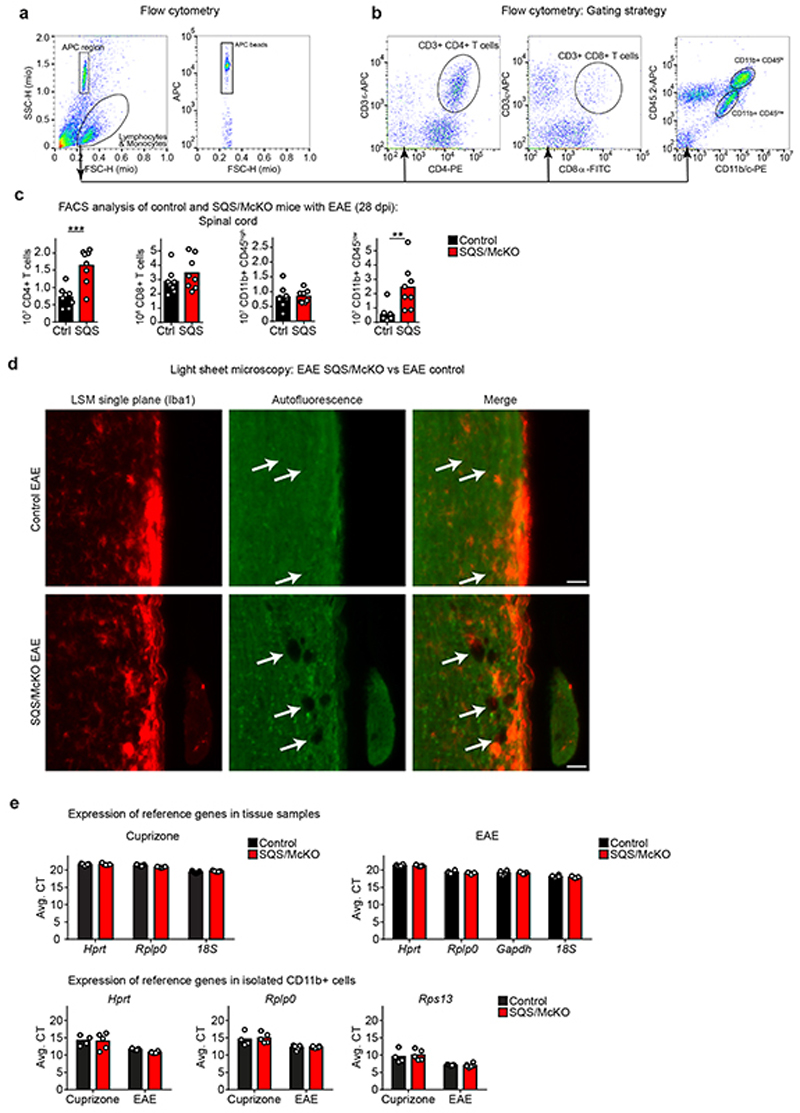
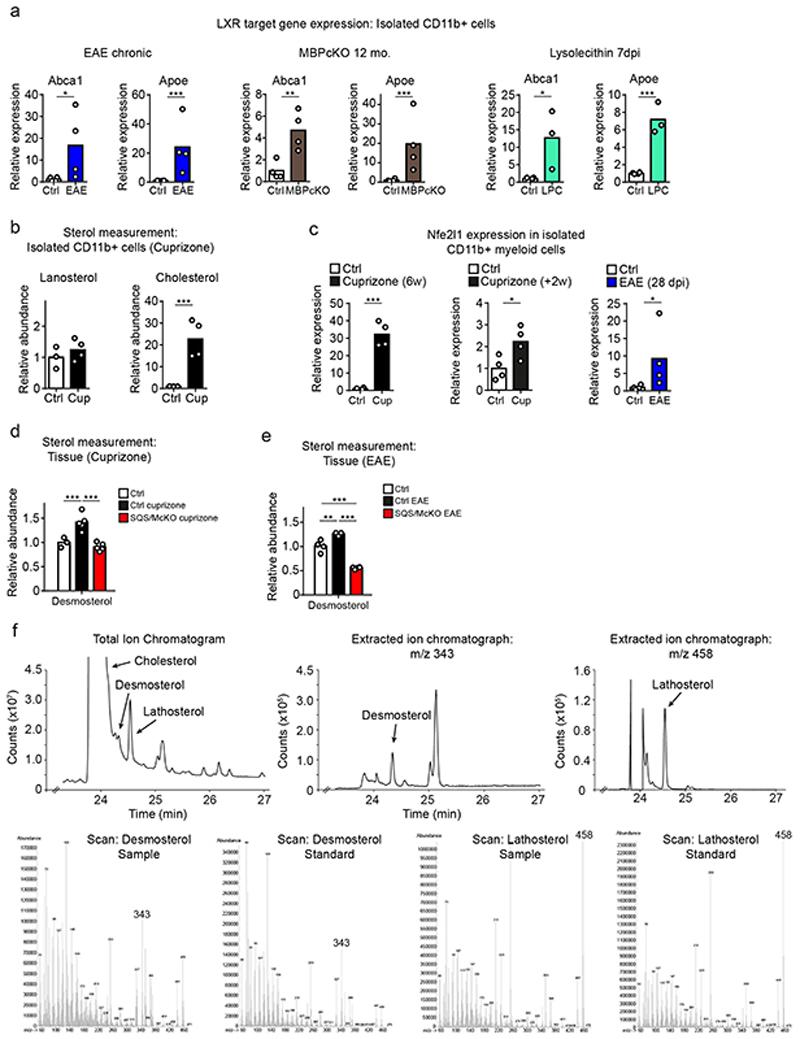
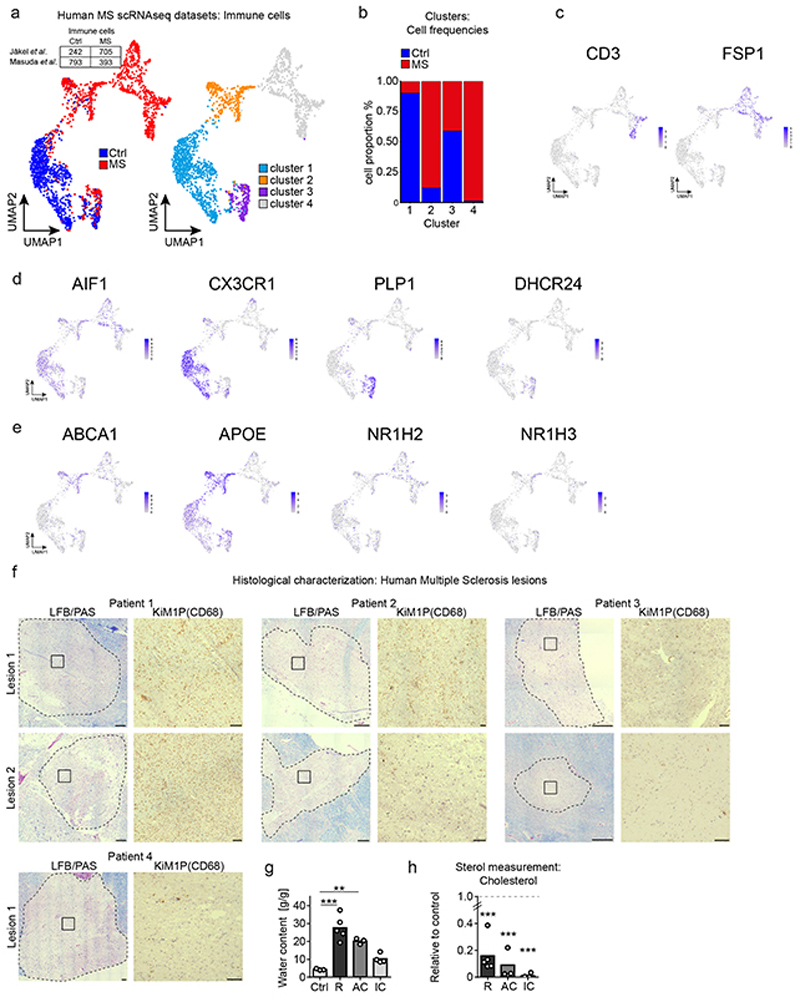
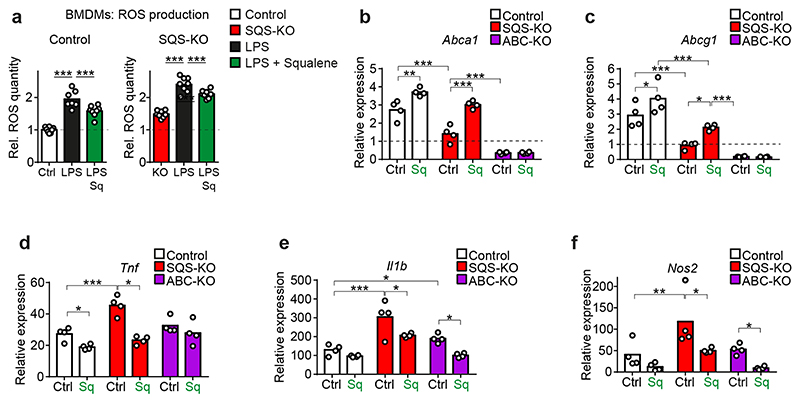
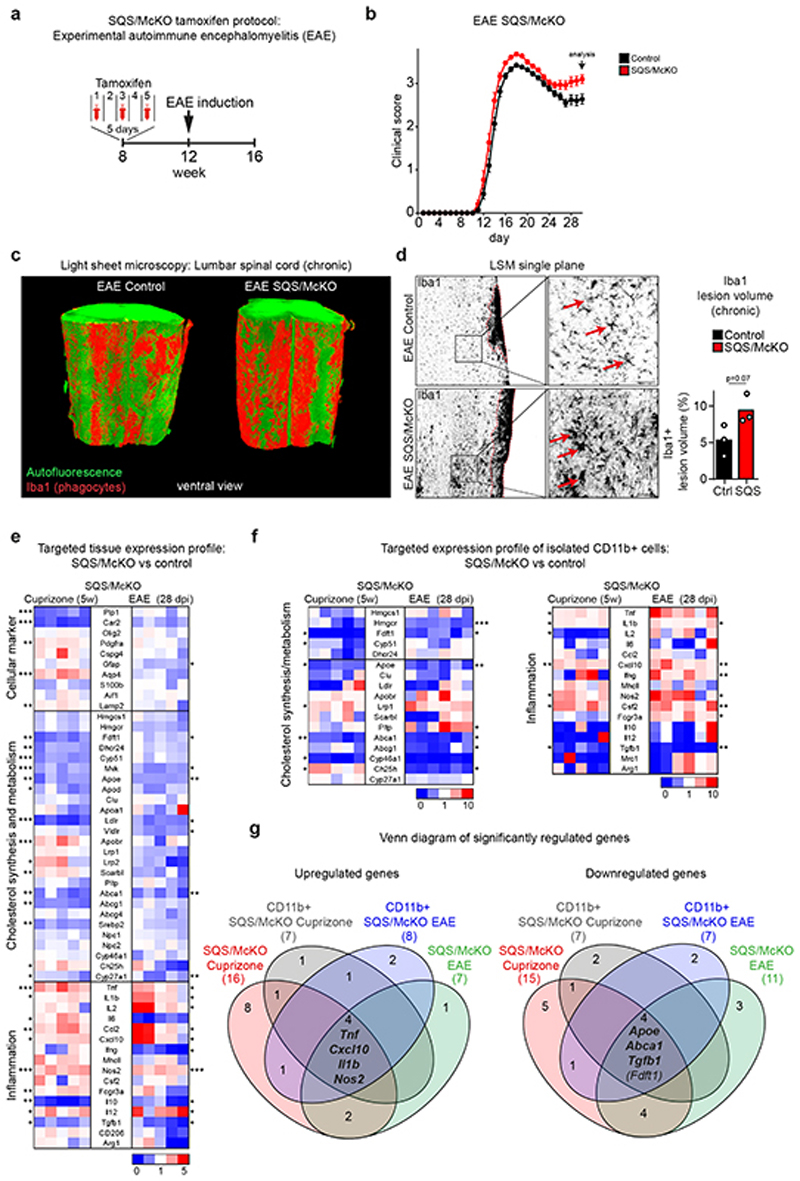
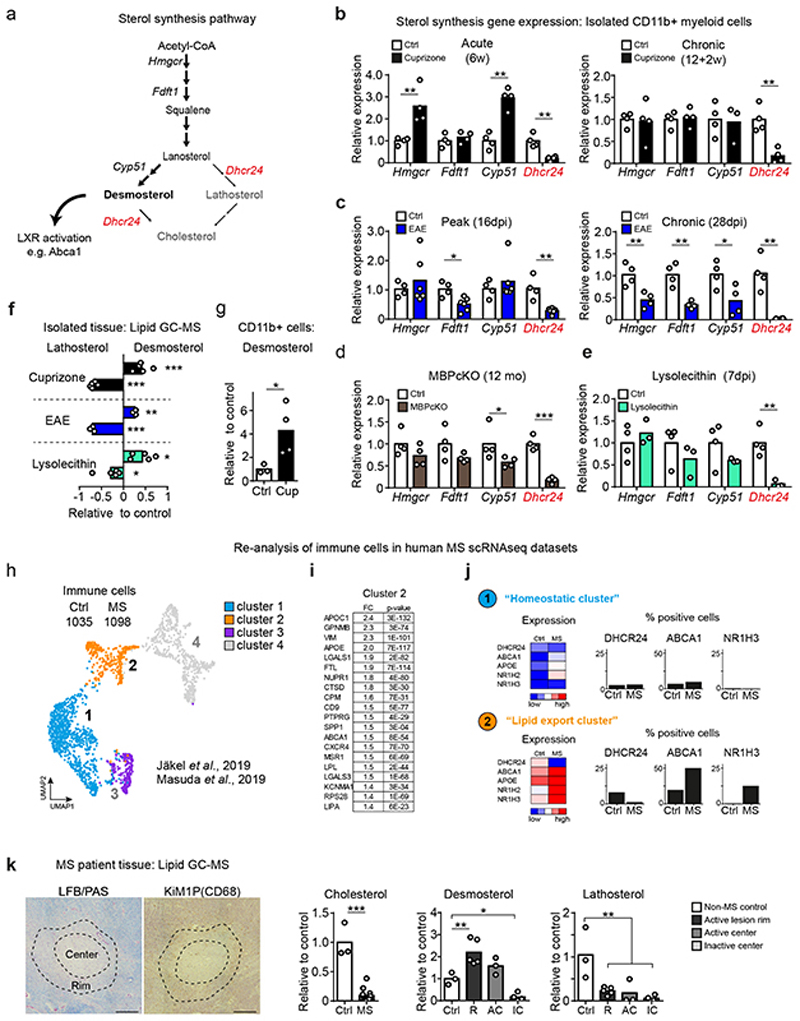
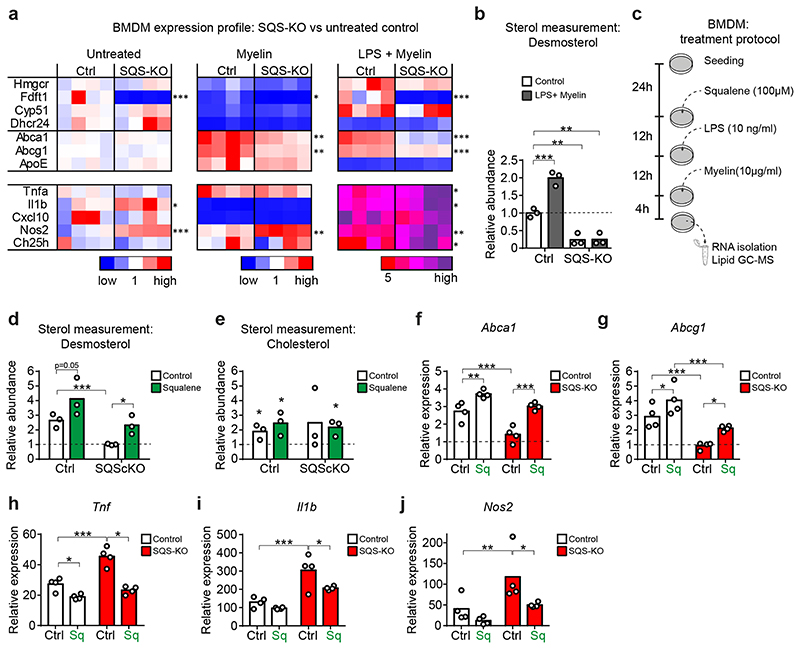
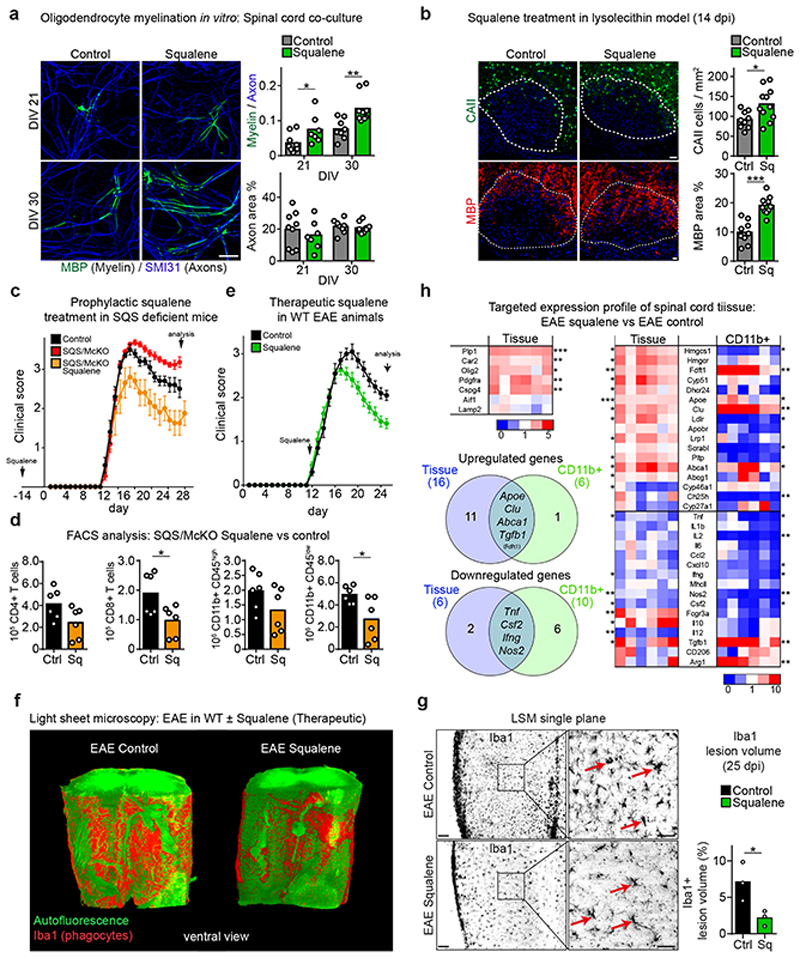
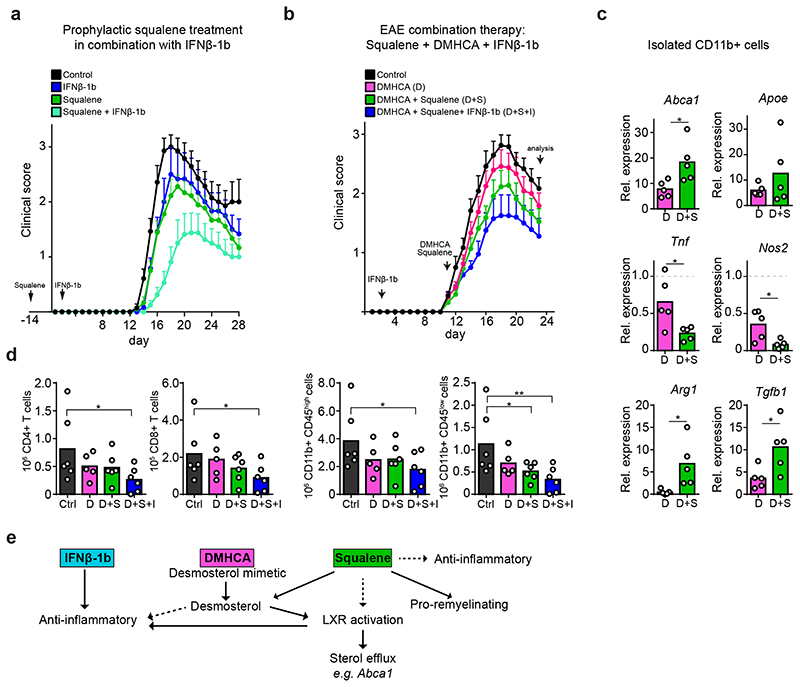
The repair of inflamed, demyelinated lesions as in multiple sclerosis (MS) necessitates the clearance of cholesterol-rich myelin debris by microglia/macrophages and the switch from a pro-inflammatory to an anti-inflammatory lesion environment. Subsequently, oligodendrocytes increase cholesterol levels as a prerequisite for synthesizing new myelin membranes. We hypothesized that lesion resolution is regulated by the fate of cholesterol from damaged myelin and oligodendroglial sterol synthesis. By integrating gene expression profiling, genetics and comprehensive phenotyping, we found that, paradoxically, sterol synthesis in myelin-phagocytosing microglia/macrophages determines the repair of acutely demyelinated lesions. Rather than producing cholesterol, microglia/macrophages synthesized desmosterol, the immediate cholesterol precursor. Desmosterol activated liver X receptor (LXR) signaling to resolve inflammation, creating a permissive environment for oligodendrocyte differentiation. Moreover, LXR target gene products facilitated the efflux of lipid and cholesterol from lipid-laden microglia/macrophages to support remyelination by oligodendrocytes. Consequently, pharmacological stimulation of sterol synthesis boosted the repair of demyelinated lesions, suggesting novel therapeutic strategies for myelin repair in MS.
Conflict of interest statement
SAB and GS are listed as inventors on pending patent claims (
Figures
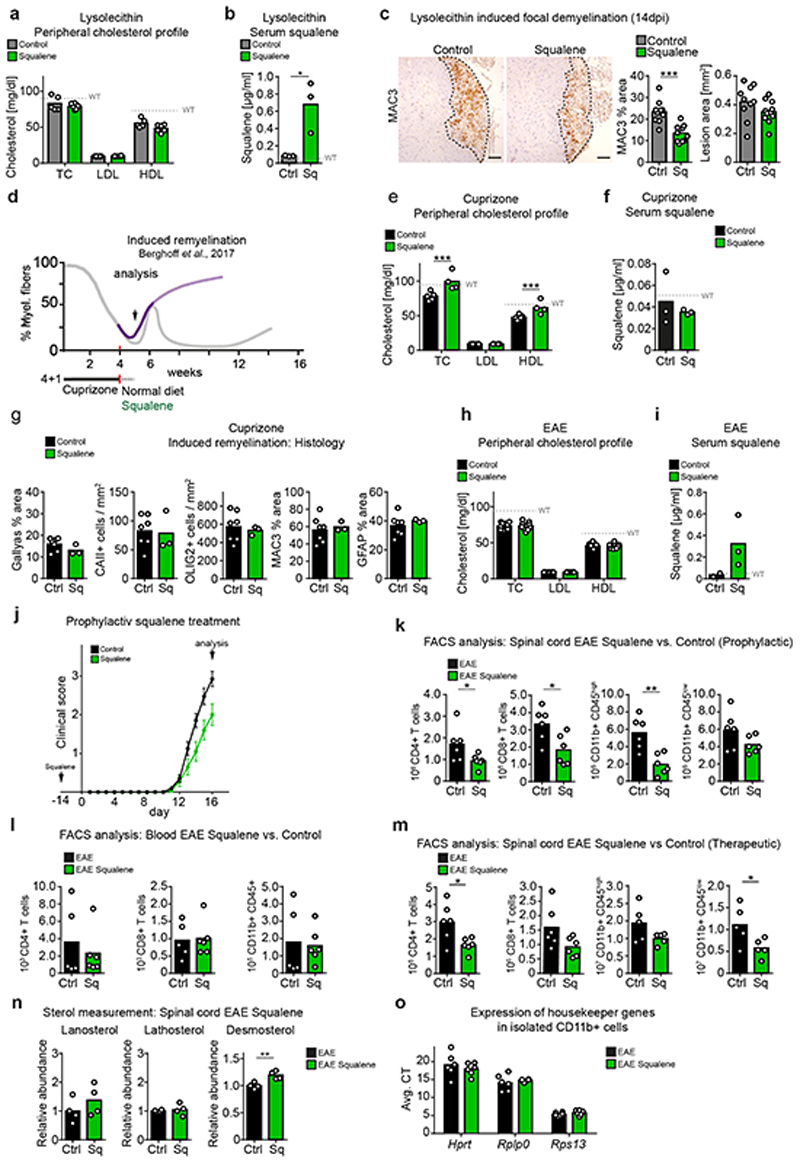
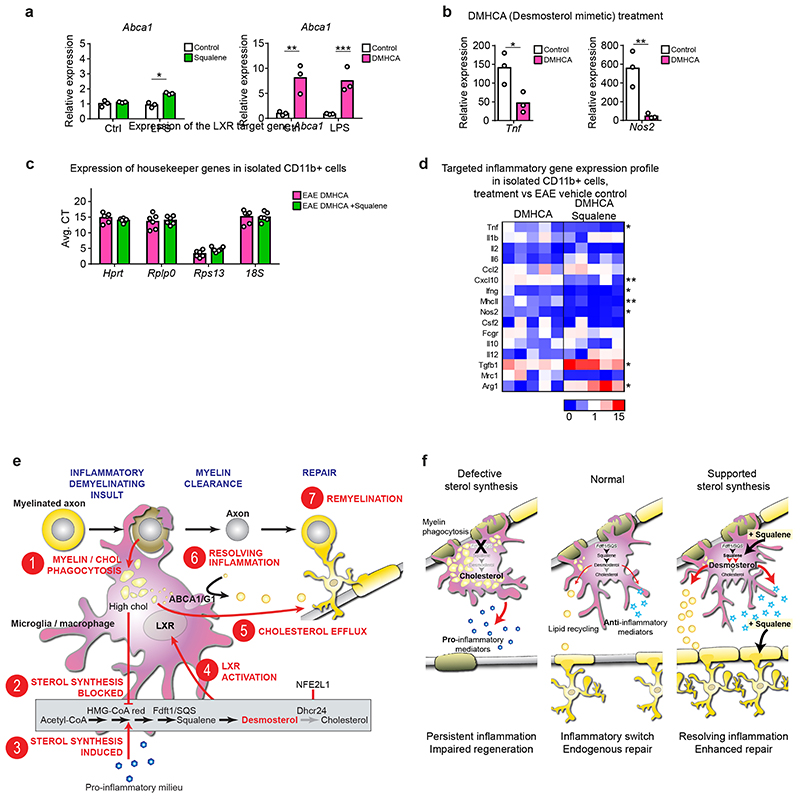
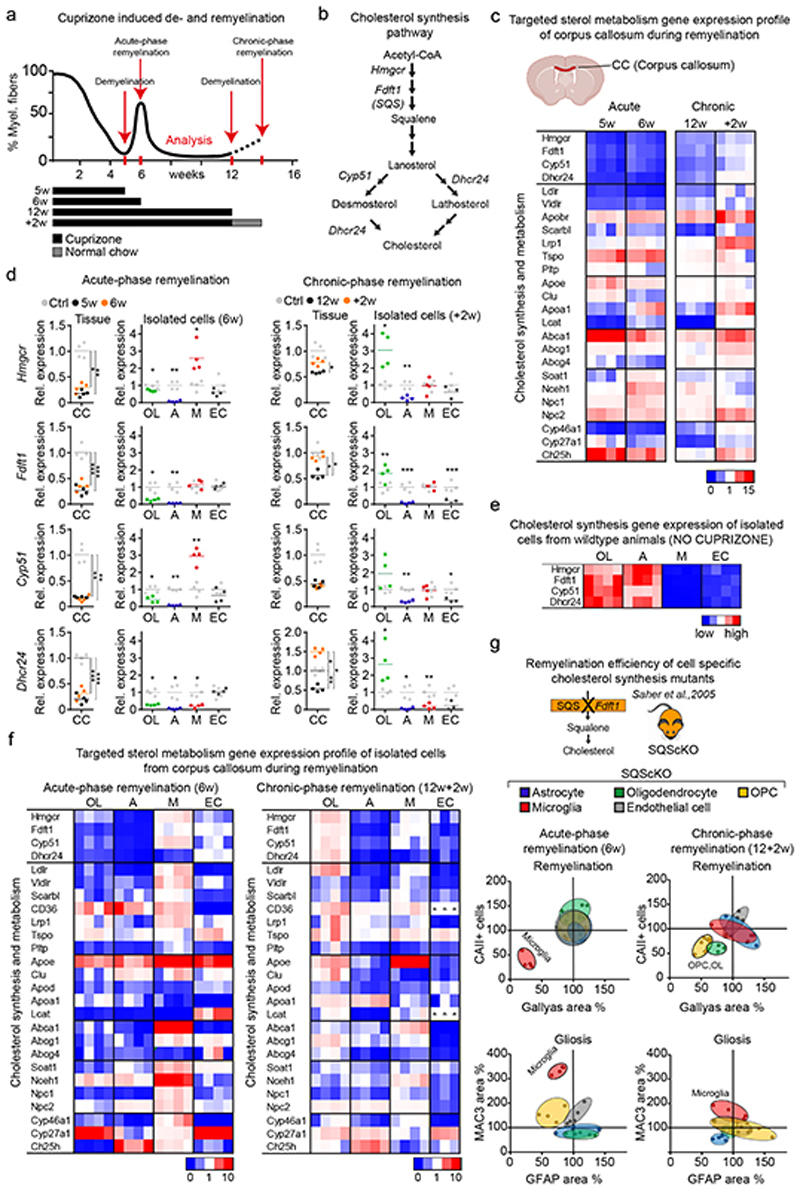
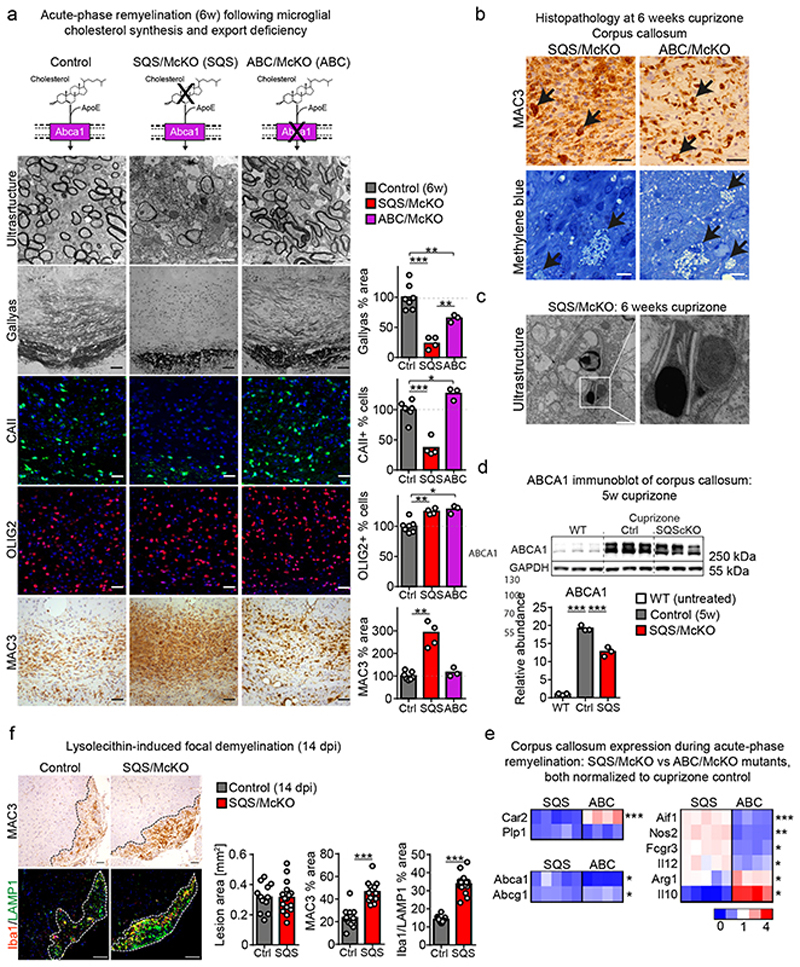
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
